
3.6: Bibliotecas genómicas y de ADNc
- Page ID
- 57257

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Aislamiento de la información genética correspondiente
En lugar de sintetizar un gen deseado, ¿podemos usar la información de aminoácidos para aislar directamente la información genética correspondiente?
- Existen dos fuentes generales de información genética:
- ADN genómico
- mRNA
Si estamos considerando el ADN genómico de eucariotas, entonces hay un par de cosas a considerar:
- La región codificante para un gen de interés puede estar interrumpida por una o más regiones intrónicas y, por lo tanto, la región codificante completa podría ser bastante larga.
- A una primera aproximación, no importa qué tejido usemos para aislar la información genómica, es decir, el contenido genómico es el mismo en todos los tejidos.
Si estamos considerando ARNm de eucariotas, podemos darnos cuenta de las siguientes ventajas:
- Los intrones se cortarán y el ARNm contendrá una región codificante contigua.
- La expresión específica de tejido de la proteína de interés puede permitirnos aislar ARNm apropiado a niveles potenciados, es decir, en tejidos donde se expresa la proteína los niveles de ARNm son considerablemente más altos que los niveles genómicos correspondientes (hay muchas más moléculas de ARNm que copias de el gen).
Bibliotecas
Una “biblioteca” es un mecanismo conveniente de almacenamiento de información genética.
- Por lo general, son información genética “genómica” o “ADNc” (es decir, ARNm en forma de ADN).
- Las secuencias genéticas deducidas de la información polipeptídica correspondiente pueden usarse para identificar información genética específica dentro de una biblioteca.
Construcción de bibliotecas de ADNc
La enzima responsable de esto es una ADN polimerasa dependiente de ARN llamada transcriptasa inversa.
- Tradicionalmente, las transcriptasas inversas se han aislado de virus cuyo genoma está realmente en forma de ARN y deben convertirse en ADN dúplex.
- Estos virus suelen llevar una transcriptasa inversa funcional junto con su componente genético de ARNm cuando infectan las células.
- Una de las transcriptasas inversas más comunes disponibles comercialmente es el virus de la leucemia murina de Moloney (MMLV).
- Esta ADN polimerasa dependiente de ARN (como todas las polimerasas) agrega nucleótidos a un polinucleótido nacente en la dirección 5' a 3' usando ARN como molde. No contiene ninguna actividad 3'->5' exonucleasa (corrección).
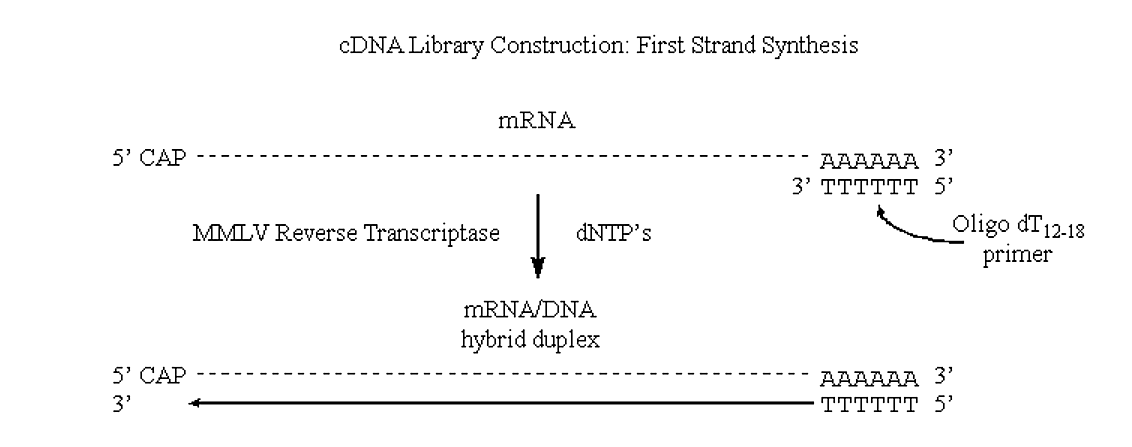
MMLV utilizará ARNm como molde, pero requiere un cebador (puede extender un cebador de ADN pero no puede sintetizar uno).
- Una de las cosas realmente interesantes de los ARNm eucariotas es la presencia de las pistas 3' poli A.
.png)
Si podemos introducir “mellas” en la mitad de ARN de este dúplex ADN/ARN entonces la situación sería muy similar a la observada en la síntesis de “hebra rezagada” del ADN genómico procariota.
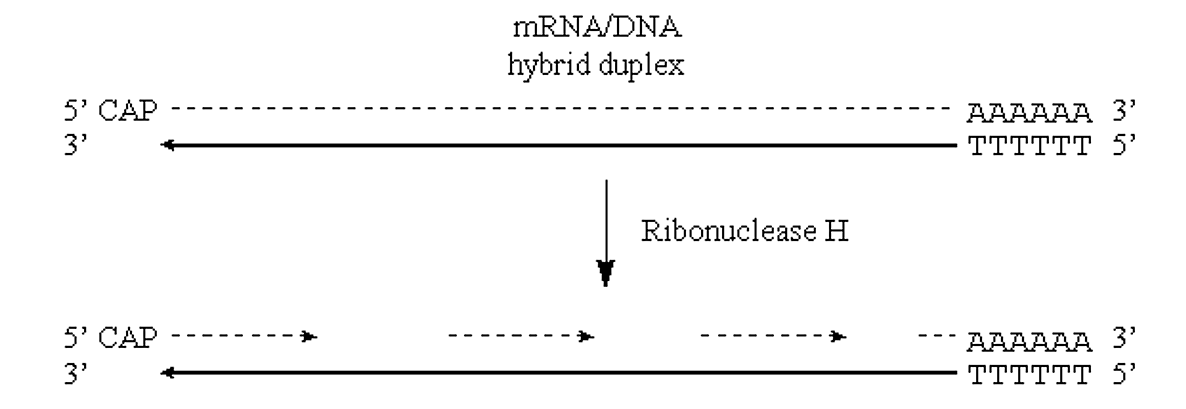
- Las mellas en la mitad de ARN de la molécula se pueden introducir a través de la acción de la enzima RNasa H.
- Esta enzima exhibe escisión endonucleolítica del resto de ARN de híbridos ARN/ADN, así como actividad 5'->3' y 3'->5' exorribonucleasa.
- Es decir, cortará el ARN y luego procederá a digerir de nuevo en ambas direcciones:
.png)
- Estos fragmentos de ARN ahora pueden servir como cebadores para la síntesis de ADN por E. coli Pol I. Esta enzima también traducirá las “mellas” para eliminar eficazmente los cebadores de ARN:
.png)
Figura 3.6.3: Síntesis de ADN
Inserción de ADNc en plásmido.
Para completar nuestra construcción de una biblioteca de ADNc útil necesitamos una forma de mantener y propagar nuestro ADNc.
- Esto se puede lograr insertando el ADNc en un plásmido apropiado.
- Hay dos formas clásicas de lograr esta hazaña:
- Colas homopolímericas
- Adición de ligadores
Colas homopolímericas
La transferasa terminal es una ADN polimerasa inusual que se encuentra solo en un tipo de célula eucariota llamada prelinfocito.
- En presencia de un catión divalente, la enzima cataliza la adición de dNTP a los extremos 3'-hidroxilo del ADN.
- Cuando el nucleótido a añadir es una purina, Mg 2+ es el catión utilizado.
- Cuando el nucleótido que se va a añadir es una pirimidina, se utiliza Co 2+.
- Dependiendo de las condiciones de reacción, se agregarán en cualquier lugar de tres a varios miles de bases.
.png)
Figura 3.6.4: Actividad de transferasa terminal
- Si cortamos nuestro plásmido y también lo tratamos con transferasa terminal, excepto que ahora agregamos la base complementaria a la que agregamos a nuestro ADNc, podemos hibridar y ligar el ADNc en el plásmido.
.png)
Figura 3.6.5: Ligación de ADNc en el plásmido
- La utilidad de insertar el inserto de ADNc de cola C en un sitio Pst I de cola G en el vector es la siguiente:
- La secuencia de reconocimiento de PstI y el sitio de escisión es
5' C T G C A G
3' G A C G T C 5' - La escisión de este sitio por PstI, seguida de cola G producirá
5' C T G C A (G) n G
3' G (G) n A C G T C 5'
Linkers
Un método alternativo para insertar fragmentos de ADNc en un vector de biblioteca es mediante la adición de “enlazadores”.
- Los enlazadores son oligonucleótidos cortos (~18 a 24 meros) que son típicamente palindrómicos y contienen una secuencia de reconocimiento de endonucleasa de restricción única o repetida.
- La naturaleza palindrómica permite que el oligonucleótido enlazador se autohibride para formar un dúplex de extremos romos.
- Si los extremos de los fragmentos de ADNc son romos, entonces el enlazador puede ligarse a ambos extremos para introducir sitios de restricción terminales útiles.
Las etapas en la adición del ligador son las siguientes:
- Tratamiento de ADNc con nucleasa S1 (para eliminar posible fragmento de ARNm de tapa 5' restante en dúplex de ADNc
- Convertir posibles extremos “irregulares” en romos mediante tratamiento con Pol I (rellenará salientes 5' y masticará voladizos 3')
- Metilar ADNc en sitios EcoRI internos potenciales mediante tratamiento con metilasa Eco RI (más S-adenosil metionina)
- Ligar los ligadores a ADNc romo metilado usando ADN ligasa T4
- Endonucleasas de corte con endonucleasa de restricción Eco RI
- Eliminar los fragmentos de conector de los fragmentos de ADNc mediante electroforesis en gel de agarosa
- Ligar el ADNc al fragmento de ADN del vector (abierto por la endonucleasa de restricción EcoRI
Nota Editorial
Este libro de texto fue publicado en 1998. El Proyecto Genoma Humano se completó en 2003.
Bibliotecas de ADN genómico
Tamaño de algunos genomas y cromosomas:
|
|
|
| (cromosoma 3 de levadura) |
|
| Genoma de Escherichia coli |
|
| El cromosoma de levadura más grande ahora mapeado |
|
| Genoma completo de levadura (completado 5/96) |
|
| Cromosoma humano más pequeño (Y) |
|
| Cromosoma humano más grande (1) |
|
| Genoma humano entero |
|
- El genoma humano contiene aproximadamente 50,000 genes únicos dentro de 3-4 mil millones de pares de bases de ADN, dispersos en 23 pares de cromosomas.
Fragmentación del ADN genómico para la construcción de bibliotecas
Digestión con endonucleasas de restricción
- Una cortadora de seis (por ejemplo, Eco RI) cortará en promedio cada 4.1 Kb. La digestión completa del ADN humano con este tipo de enzima dará como resultado aproximadamente 1 x 10 6 fragmentos únicos.
- ¿Cuál es la probabilidad de encontrar un clon dentro de una biblioteca dada?
La probabilidad exacta de tener cualquier secuencia de ADN dada en la biblioteca se puede calcular a partir de la ecuación
Por ejemplo, ¿qué tan grande necesitaría una biblioteca (es decir, cuántos clones) para tener una probabilidad del 99% de encontrar una secuencia deseada representada en una biblioteca creada por digestión con un cortador de 6?
Así, a partir de este tipo de análisis podemos ver que necesitamos una tecnología que nos permita lograr lo siguiente:
- Inserción estable de fragmentos de ADN relativamente grandes en nuestro vector de clonación
- Alta eficiencia de inserción y la capacidad de manejar grandes cantidades de clones
- Por ejemplo, al sembrar colonias de E. coli en una placa Petri de 3", la densidad práctica máxima para permitir el aislamiento de colonias individuales es de aproximadamente 100-200 colonias por placa.
- Si tuviéramos que tratar de platicar nuestra biblioteca de 3.37 x 10 6 de tal manera necesitaríamos alrededor de 22,500 platos.
- No solo eso, sino que fragmentos de ADN tan grandes no son bien tolerados en vectores de clonación típicos de E. coli como pBR322.
Los vectores lambda de bacteriófagos se utilizan comúnmente para la construcción de bibliotecas genómicas
El bacteriófago l es un fago de E. coli con un tipo de partícula de fago icosaédrico que contiene el genoma viral:
.png)
Figura 3.6.6: Bacteriofase l
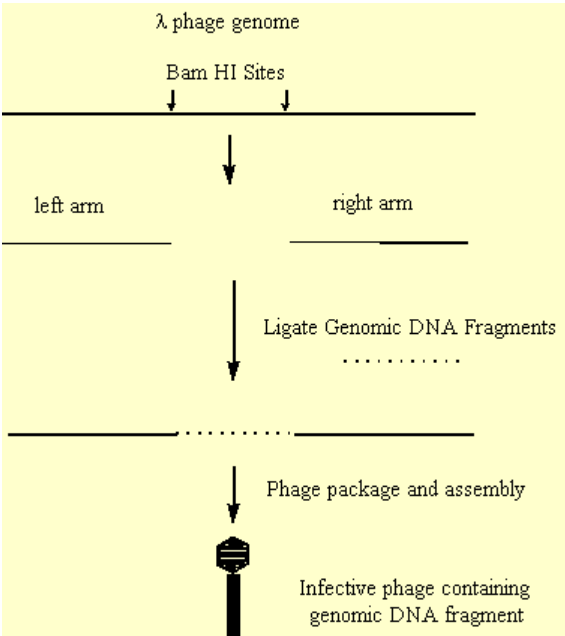
- Durante la replicación, el ADN del fago se produce en una forma concatamérica, que se escinde mediante endonucleasas apropiadas para permitir el empaquetamiento de un solo genoma dentro de la cápside del fago.
- Se encontró que las regiones internas del genoma del fago, que no eran esenciales para la replicación del fago, podían eliminarse y reemplazarse con ADN de interés.
- Este ADN híbrido podría empaquetarse de manera eficiente y formar un fago infeccioso.
.png)
Figura 3.6.7: Creación de fagos ineficaces
Las ventajas de este tipo de sistemas frente a plásmidos como pBR322 son:
- El genoma del fago es capaz de empaquetarse eficientemente con insertos de ADN de hasta 20 Kb.
- Además, los fagos empaquetados son altamente infecciosos e infectan E. coli con una eficiencia mucho mayor que los métodos de transformación plasmídica.
La digestión incompleta del ADN genómico permitirá la identificación de solapamientos de secuencias
La digestión completa con una endonucleasa dará como resultado una biblioteca que no contiene fragmentos superpuestos:
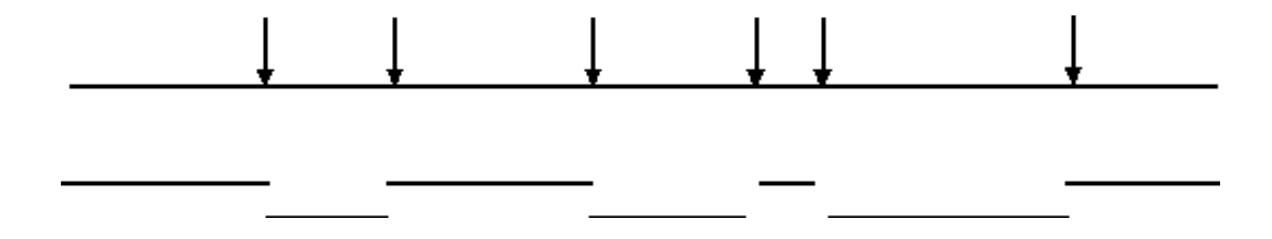
Sin embargo, la digestión incompleta dará como resultado una biblioteca que contiene fragmentos superpuestos:
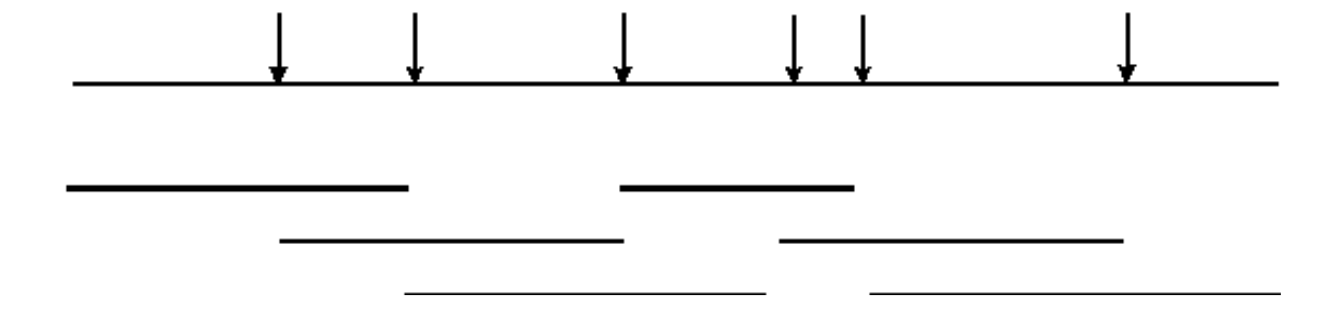
- Así, la información de secuencia obtenida de un clon permitirá el aislamiento de clones que contienen información de secuencia vecina (solapada).
- Esto puede permitir que se obtengan grandes tramos contiguos de información de secuencia (” Caminata Cromosómica “).
Bibliotecas de sondeo
Una vez construida una biblioteca (ADNc o genómica) queremos ser capaces de identificar clones que contengan ADN de interés.
- Por ejemplo, a partir de la información de la secuencia proteica podemos deducir posibles tramos de la secuencia de ADN correspondiente (sin embargo habrá ambigüedad debido a la degeneración de los codones).
- Si podemos sintetizar un oligonucleótido complementario a nuestra secuencia de ADN de interés, podemos usarlo para hibridar específicamente con el clon apropiado en nuestra biblioteca (es decir, para sondear nuestra biblioteca).
En metodologías estándar el oligonucleótido se fosforila en el extremo 5' con P-ATP g 32 radiomarcado y polinucleótido quinasa T4.
- La sonda se incuba entonces con placas de fago individuales que se han fijado sobre nitrocelulosa y su ADN desnaturalizado por tratamiento con base.
- Si la placa contiene ADN complementario a la secuencia de la sonda, la sonda hibridará.
- Si la nitrocelulosa (que contiene muchas placas individuales) se expone a una película de rayos X, solo las placas con sonda hibridada se mostrarán (como una mancha oscura):
.png)
Figura 3.6.8: Placa radiomarcada
Falsos positivos
Si estamos diseñando sondas de ADN a partir de información de secuencias proteicas tendremos posible ambigüedad en nuestra secuencia de ADN deducida utilizada para el diseño de la sonda.
- Por lo general, se utilizan oligonucleótidos 14-24mer como sondas, una sonda 14-24mer significa que necesitamos un tramo de 5-8 aminoácidos en el polipéptido.
- Dada la elección, las mejores secuencias de aminoácidos a buscar en un polipéptido son aquellas con baja degeneración de codones (ver arriba).
- Así, buscaríamos un tramo corto de secuencia polipeptídica ojalá que contenga Met o Trp, y con los aminoácidos restantes que comprenden Phe, Tyr, His, Gln, Asn, Lys, Asp, Glu o Cys.
- Se deben evitar regiones incluyendo Leu, Arg o Ser (6 codones cada una).
Durante la síntesis de oligonucleótidos se incorporarán múltiples bases en posiciones ambiguas.
- Así, nuestra sonda será en realidad una mezcla de oligonucleótidos.
- Cuanto mayor sea la degeneración, mayor será la posibilidad de los “falsos positivos”, es decir, clones que hibridan pero no están relacionados con la secuencia real que queremos.
- Los clones positivos se secuencian y la secuencia de aminoácidos deducida se compara con la información de la secuencia de nuestros polipéptidos para identificar los clones correctos.
Anticuerpos (Inmunoglobulinas)
Si el vector particular, o fago, usado para construir una biblioteca de ADNc contiene una región promotora aguas arriba del sitio de inserción, podemos seleccionar los clones deseados buscando la expresión de la proteína de interés.
- En este caso, necesitamos un ensayo que sea tanto sensible (no vamos a estar produciendo mucha proteína) como específico (queremos minimizar cualquier falso positivo).
- Uno de los mejores ensayos, que es tanto sensible como específico, hace uso de anticuerpos.
Antígeno, anticuerpo, epítopo
Uno de los mecanismos de defensa de los vertebrados es la capacidad de distinguir entre auto y no-auto moléculas.
- Así, si una molécula extraña (ya sea de otra especie o a veces de otro individuo dentro de una especie) invade un organismo vertebrado, el sistema inmune funciona para aprender a identificar esa molécula.
- En futuras invasiones por la misma molécula, el organismo monta una defensa contra ella produciendo anticuerpos específicos que reconocen y se unen al antígeno extraño.
- Cuando los anticuerpos se unen al antígeno ciertos glóbulos blancos (macrófagos y monocitos) reconocen al cuerpo invasor como extraño y responden destruyéndolo.
Los anticuerpos son moléculas en forma de 'Y' que contienen dos cadenas pesadas idénticas y dos cadenas ligeras idénticas.
- El tallo de la 'Y' comprende el dominio Fc (constante), y los 'brazos' de la 'Y' comprenden los dominios Fab (variable).
- Los antígenos se unen a las regiones determinantes de complementariedad (CDR) localizadas en los extremos de los dominios Fab.
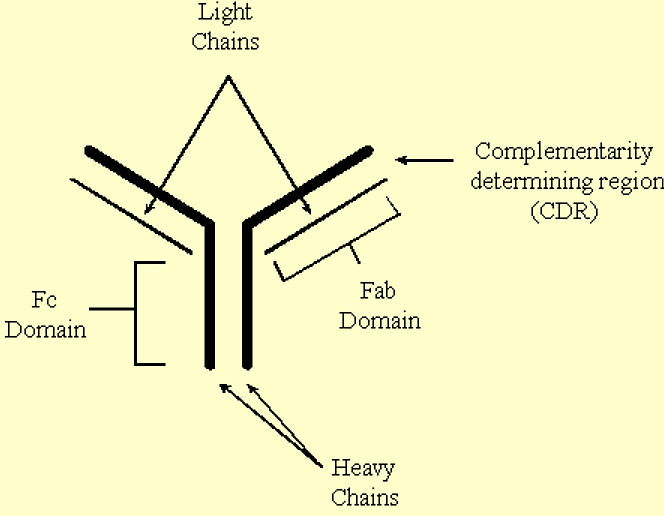.png)
Figura 3.6.9: Estructura de anticuerpos
Los anticuerpos son sintetizados por los linfocitos B. Cada linfocito B es capaz de producir un único tipo de anticuerpo dirigido contra un determinante estructural específico, o epítopo, sobre un antígeno.
- Así, una respuesta inmune a un antígeno proteico puede resultar en una población de linfocitos B produciendo cada uno anticuerpos que reconocen un determinante estructural diferente de la proteína extraña.
- Un epítopo puede ser una región contigua de 5 ó 6 aminoácidos en el polipéptido extraño, o el epítopo puede comprender media docena de aminoácidos llevados en yuxtaposición en la proteína nativa, pero ampliamente espaciados en la secuencia polipeptídica.
- Así, algunos anticuerpos reconocerán igualmente bien las formas nativas y desnaturalizadas de una proteína extraña, mientras que otros anticuerpos solo pueden reconocer una u otra.
Si la proteína de interés ha sido purificada puede ser utilizada para inducir una respuesta inmune en un animal huésped.
- Los animales hospedadores típicos incluyen ratón, pollo, conejo, cabra, oveja, caballo y ocasionalmente, humanos.
- Después de una inmunización inicial, seguida de una o más inyecciones de refuerzo, los linfocitos B del animal huésped pueden producir anticuerpos dirigidos contra el antígeno.
- Los anticuerpos pueden ser purificados a partir de muestras de sangre extraídas del animal. Se dice que tales preparaciones de anticuerpos son policlonales.
- Esto se refiere al hecho de que los anticuerpos presentes son de una colección de diferentes linfocitos B y así reconocerán una variedad de epítopos diferentes en la proteína antigénica.
- La capacidad de aislar anticuerpos de muestras de sangre significa que el animal huésped no necesita ser destruido.
- Por supuesto, el tamaño del animal determina la cantidad de anticuerpos que se pueden obtener. Por ejemplo, un conejo puede aportar 5 ml de sangre cada dos semanas, un ratón aporta significativamente menos, mientras que un caballo puede aportar bastante más.
Un antibodiy aislado de una sola población de linfocitos B se denomina monoclonal.
- Reconoce un único epítopo sobre la proteína antigénica.
- Los linfocitos B productores de anticuerpos pueden aislarse del bazo o de los ganglios linfáticos. Sin embargo, tienen una vida finita en cultivo, es decir, sufrirán cierto número de divisiones celulares y luego morirán.
- Estas células pueden, sin embargo, fusionarse con linfocitos inmortales (mieloma canceroso) para producir una célula de hibridoma.
- Tal célula es inmortal como el mieloma, y produce un anticuerpo específico a partir del linfocito B. La capacidad de crecer indefinidamente en cultivo permite el aislamiento de cantidades útiles de anticuerpos monoclonales específicos.
A veces inmunizar con la proteína de interés es problemático: no se pueden producir cantidades apropiadas de material purificado, o la proteína es tóxica en sí misma al nivel de dosificación necesario para producir una respuesta inmune.
- Si se conoce información de secuencia parcial, entonces grandes cantidades de polipéptidos que representan fragmentos cortos de la proteína, pueden sintetizarse y usarse para inmunizar al animal.
- A menudo, estos polipéptidos se unen covalentemente a una proteína portadora (típicamente albúmina sérica) para potenciar la respuesta antigénica.
- Los anticuerpos producidos contra tales péptidos reconocerán solo epítopos dentro del polipéptido. Así, incluso los anticuerpos policlonales estarían bastante limitados en su reconocimiento de epítopos.
Al igual que con los oligonucleótidos radiomarcados, se pueden usar anticuerpos para identificar clones de biblioteca que contienen un ADNc de interés. Este método, por supuesto, se basaría en un vector huésped o fago que contiene un promotor aguas arriba del sitio de inserción del ADN genómico.
- Los anticuerpos se pueden usar para cribar placas virales o clonias plasmídicas que se han unido a nitrocelulosa.
- Los anticuerpos unidos se pueden identificar usando proteína A radiomarcada (que se une a inmunoglobulinas) o a través de un segundo anticuerpo (que, como la proteína A, puede reconocer inmunoglobulinas generales) que tiene un colorante o enzima liberadora de colorante unido covalentemente.