
9.5: Impacto de los estados de enfermedad y daño neural en el control motor
- Page ID
- 148294
- Describir los síntomas generales de los principales trastornos motores del sistema nervioso
- Categorizar los trastornos motores en función de sus mecanismos neuroantómicos o neuroquímicos subyacentes
- Explicar algunos de los tratamientos para algunos de los trastornos mayores
Los trastornos motores y sus mecanismos neuroanatómicos/neuroquímicos
Dado que el control motor implica una colección tan extensa de estructuras y circuitos en todo el sistema nervioso central y periférico, es sorprendente lo bien que funcionan y, a menudo sin esfuerzo, estos sistemas funcionan cada segundo de nuestras vidas. Sin embargo, la naturaleza integral de los sistemas motores también permite múltiples vías de daño potencial o degeneración que pueden llevar a profundas consecuencias en la capacidad de uno para moverse o incluso sobrevivir.
El daño o enfermedad puede ocurrir en regiones del prosencéfalo asociadas con Neuronas Motrices Superiores en la corteza o los ganglios basales, y/o regiones de la Neurona Motora Inferior de la médula o médula espinal. Estas distinciones anatómicas pueden ser críticas en el diagnóstico y tratamiento de trastornos motores específicos.
Esclerosis Múltiple
La esclerosis múltiple (EM) es una enfermedad motora progresiva que ataca a las neuronas tanto en el sistema nervioso central como periférico. El modo principal de este ataque es la destrucción del recubrimiento de mielina alrededor del axón, lo que finalmente ralentizará la conductancia potencial de acción. A medida que los axones se desmielinizan, se forman parches inflamatorios llamados lesiones. A medida que avanza la enfermedad, se destruyen los oligodendrocitos productores de mielina y, en última instancia, los propios axones. Existe evidencia convincente de que la destrucción es causada por la activación selectiva del sistema inmune celular y las moléculas inflamatorias, apoyando así la idea de que la EM es una enfermedad autoinmune.
Los hallazgos de estudios de MRI indican que un individuo con EM puede tener anomalías alrededor de sitios como el ventrículo lateral, nervio óptico, tronco encefálico, médula espinal, cerebelo y otras áreas (ver Figura\(\PageIndex{1}\) a continuación).
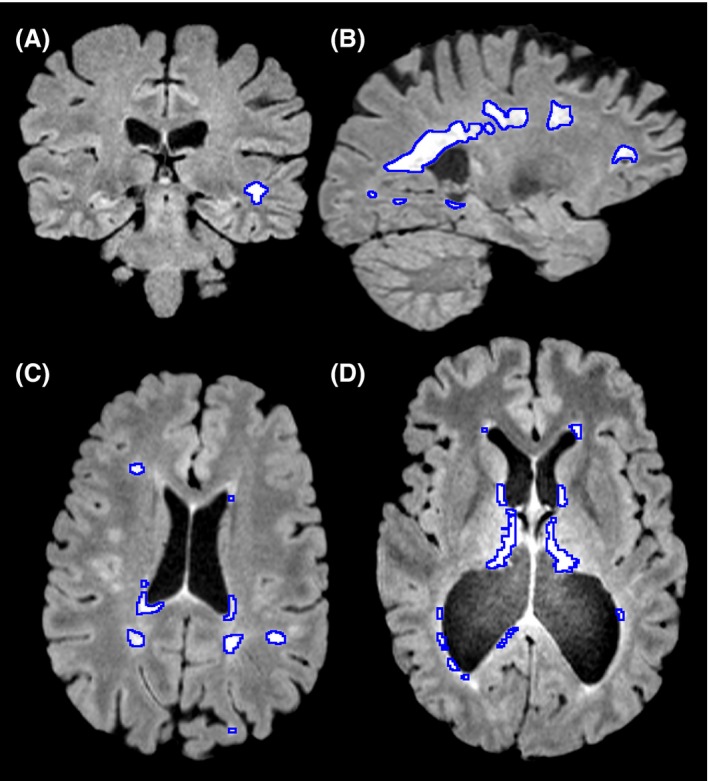
Síntomas de EM
La esclerosis múltiple también se presenta de manera diferente en las fases aguda y crónica. Durante la fase aguda, el padecimiento se asocia con síntomas intermitentes, mientras que la fase crónica se asocia con formas progresivas de la enfermedad y aumento de la severidad de los síntomas.
La mayoría de las personas experimentan sus primeros síntomas de EM entre los 20 y 40 años con visión inicial borrosa o doble, distorsión del color rojo-verde o incluso ceguera en un ojo. La mayoría de los pacientes con EM también experimentan debilidad muscular en sus extremidades y dificultad con la coordinación y el equilibrio. Estos síntomas pueden ser lo suficientemente graves como para perjudicar el caminar o incluso pararse. En el peor de los casos, la EM puede producir parálisis parcial o completa.
La mayoría de las personas con EM también exhiben parestesias, sentimientos sensoriales anormales transitorios como entumecimiento, punzadas o sensaciones de “alfileres y agujas”. Algunos también pueden experimentar dolor. Los impedimentos del habla, temblores y mareos son otras quejas frecuentes. Ocasionalmente, las personas con EM tienen pérdida auditiva. Aproximadamente la mitad de todas las personas con EM experimentan deficiencias cognitivas como dificultades con la concentración, la atención, la memoria y el mal juicio, pero estos síntomas suelen ser leves y frecuentemente se pasan por alto. La depresión es otra característica común de la EM.
Causas potenciales de la EM
Actualmente, no hay una causa conocida para la EM aunque se han planteado muchas hipótesis diferentes. Sorprendentemente, no se conocen genes asociativos que hayan sido implicados en causar EM, y los científicos creen que podría haber una interacción compleja entre genes y factores ambientales que producen desmielinización. Las teorías más comunes sobre la (s) causa (s) de la esclerosis múltiple incluyen:
- Infección viral que resulta en una reacción autoinmune
- Factores genéticos: predisposición hereditaria posiblemente a través del sistema inmune, aunque no se ha descubierto una mutación en un gen
- Factor (es) ambiental (es) que podrían trabajar con la genética como los niveles bajos de vitamina D o el tabaquismo.
A pesar de que se trata de un trastorno bien caracterizado, todavía no sabemos mucho sobre sus causas.
Enfermedad de Parkinson
La enfermedad de Parkinson (EP) pertenece a un grupo de afecciones llamadas trastornos del sistema motor, que provocan movimientos involuntarios o incontrolables del cuerpo. Se desconoce la causa precisa de la EP, pero algunos casos son hereditarios mientras que otros se consideran a partir de una combinación de factores genéticos y ambientales que desencadenan la enfermedad. En la EP, las células cerebrales se dañan o mueren en la sustancia negra que produce el neurotransmisor dopamina, una sustancia química necesaria para producir movimientos suaves y decididos.
Síntomas de la EP
Los cuatro síntomas principales de la EP son:
- temblor—temblor que tiene un movimiento rítmico característico de ida y vuelta
- rigidez: rigidez muscular o resistencia al movimiento, donde los músculos permanecen constantemente tensos y contraídos
- bradicinesia—ralentización del movimiento espontáneo y automático que puede dificultar la realización de tareas simples o realizar movimientos de rutina rápidamente
- inestabilidad postural: alteración del equilibrio y cambios en la postura que pueden aumentar el riesgo de caídas.
Otros síntomas pueden incluir dificultad para tragar, masticar o hablar; cambios emocionales; problemas urinarios o estreñimiento; demencia u otros problemas cognitivos; fatiga; y problemas para dormir.
La EP suele afectar a personas de alrededor de los 70 años pero puede ocurrir antes. La EP afecta más a hombres que a mujeres. Actualmente no existen pruebas específicas que diagnostiquen la EP esporádica.
Posibles causas de la EP
El modelo actual para la mayoría de las condiciones neurodegenerativas es que las neuronas mueren a través de la apoptosis, una forma programada de muerte celular. Aunque este tipo de muerte celular ocurre durante el desarrollo normal, se cree que las enfermedades neurodegenerativas como la Enfermedad de Parkinson y la Enfermedad de Alzheimer utilizan vías apoptóticas. Esto sugiere que existe una proteína componente que se pliega aberrantemente para producir apoptosis. Esto tiene implicaciones clínicas muy importantes ya que activar o desactivar genes/proteínas específicos es un método para detener o posiblemente revertir la neurodegeneración.
La mayoría de los casos de este trastorno parecen ser esporádicos (es decir, de origen no genético) aunque muy pocos casos parecen tener un origen genético. La enfermedad de Parkinson se asocia con mayor frecuencia con la pérdida de núcleos “pigmentados” en el cerebro y generalmente implica la pérdida de un grupo de neuronas que se encuentran en la sustancia negra (ver Figura\(\PageIndex{2}\)). Las neuronas de la sustancia nigra son dopaminérgicas y pigmentadas porque contienen la proteína melanina.
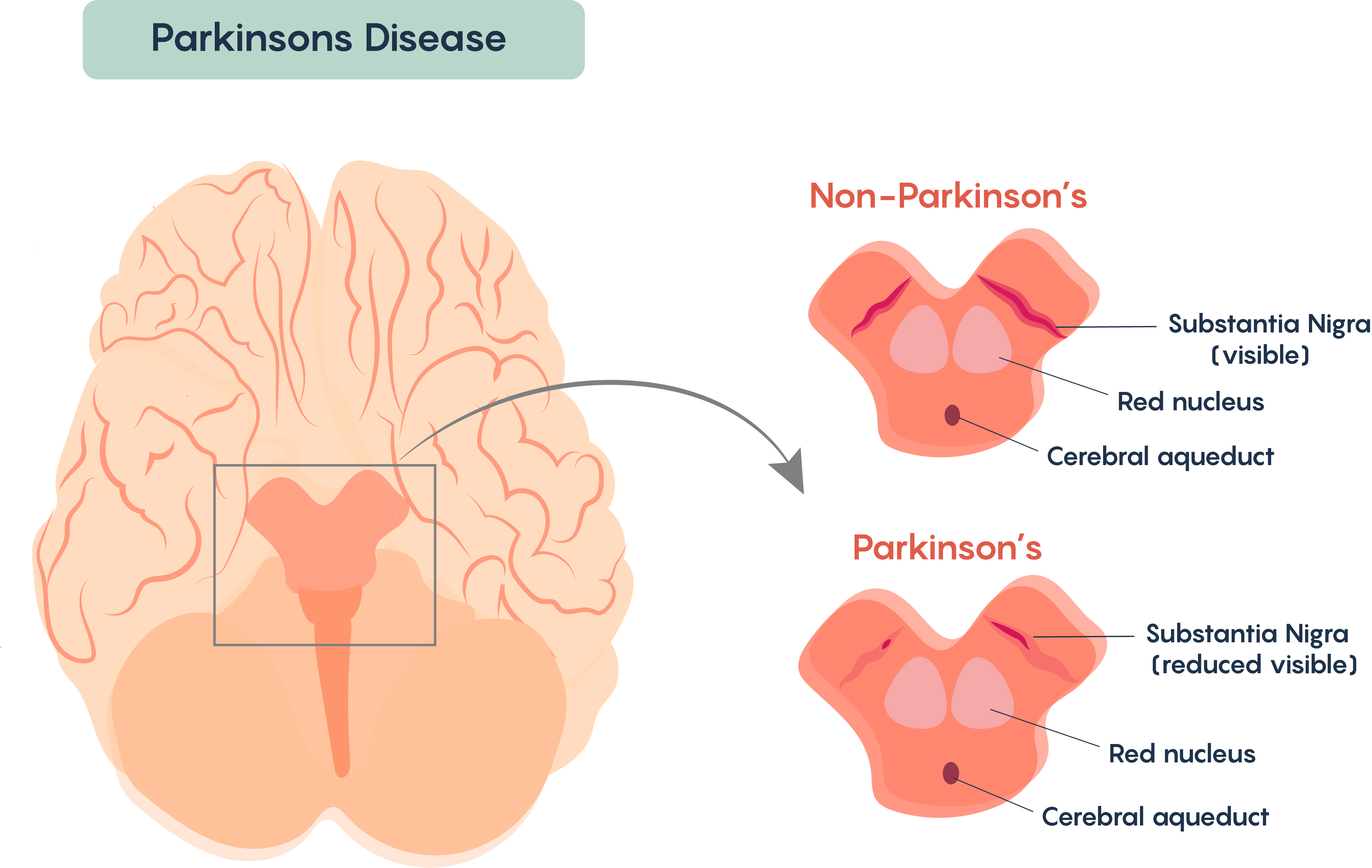
La pérdida de las células de la sustancia nigra afecta el procesamiento y ejecución del movimiento voluntario en individuos con enfermedad de Parkinson. Similar a la EM, una vez diagnosticados, los síntomas se vuelven continuos y progresivos, es decir, los síntomas empeoran con el tiempo. Nuevamente, hay muchas similitudes con la EM, y no se conoce una cura para la Enfermedad de Parkinson. La enfermedad sigue siendo idiopática aunque existen algunas causas conocidas de la enfermedad de Parkinson, incluyendo pérdida de movimiento tras aterosclerosis cerebral, encefalitis viral, y como resultado de efectos secundarios de fármacos como fenotiazidas y reserpina.
La alfa-sinucleína es una proteína natural dentro de las neuronas. Las mutaciones en los genes PARK1 y PARK4 que normalmente codifican para alfa-sinucleína se han asociado con la enfermedad de Parkinson. Como tal, muchos modelos animales de la Enfermedad de Parkinson buscan la producción de la forma fibrilar de la alfa-sinucleína a medida que se despliega y luego se acumula dentro de las neuronas de la sustancia negra conocidas como cuerpos de Lewy (ver Figura\(\PageIndex{3}\)). Se ha planteado la hipótesis de que el mal plegamiento y acumulación de alfa-sinucleína es la razón por la que las neuronas sufren apoptosis, aunque el mecanismo exacto de cómo ocurre esto aún no se ha aclarado.
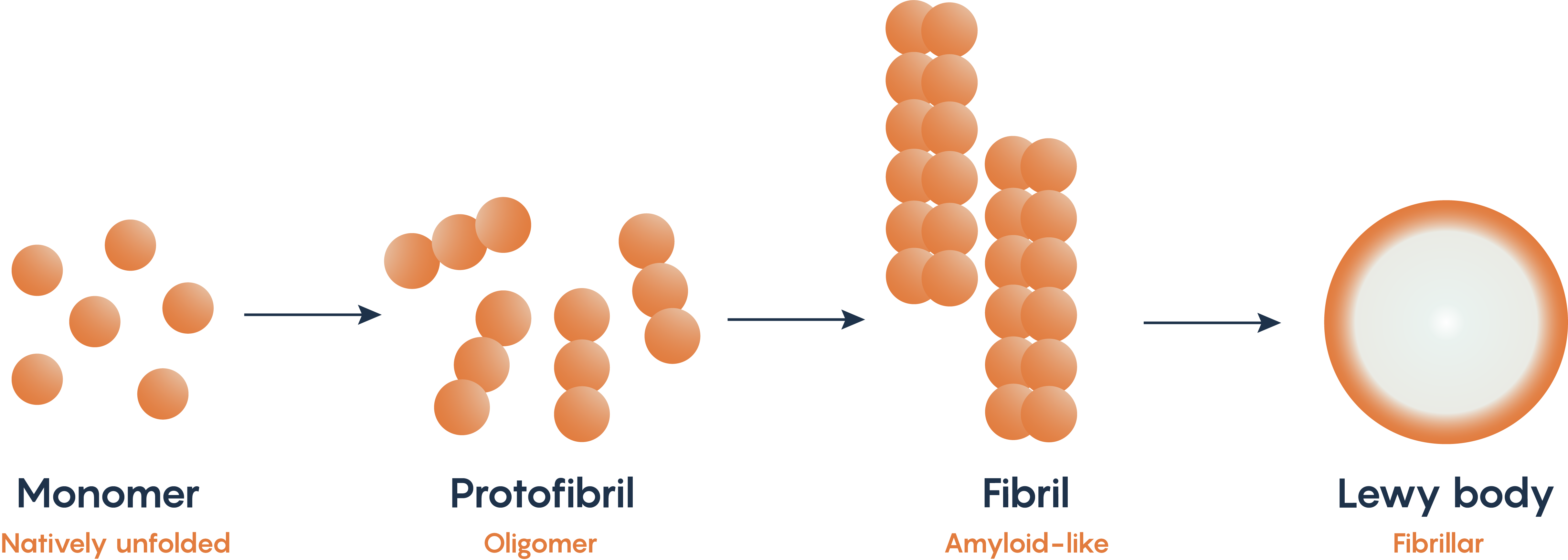
Tratamiento de la EP
En la actualidad, no hay cura para la EP, pero una variedad de medicamentos proporcionan un alivio dramático de los síntomas. Por lo general, a los individuos afectados se les administra levodopa (l-dopa) combinada con carbidopa. La carbidopa retrasa la conversión de levodopa en dopamina hasta que llega al cerebro. Las células nerviosas pueden usar levodopa para producir dopamina y reponer el suministro cada vez menor del cerebro. Aunque la levodopa ayuda a la mayoría de las personas con EP, no todos responden por igual a la droga. Los síntomas de bradicinesia y rigidez responden mejor, mientras que el temblor puede reducirse solo marginalmente. Es posible que los problemas de equilibrio y otros síntomas no se alivian en absoluto.
Existe una gran variedad de otros medicamentos que afectan el sistema de dopamina y tratan los síntomas de la EP. Algunos imitan el papel de la dopamina en el cerebro, haciendo que las células nerviosas reaccionen como lo harían a la dopamina, mientras que otros prolongan los efectos de la levodopa al prevenir la descomposición de la dopamina en el cerebro. Además de estos fármacos dopaminérgicos, se ha demostrado que los fármacos anticolinérgicos ayudan a controlar los síntomas de temblor y rigidez.
En algunos casos, la cirugía puede ser apropiada si la enfermedad no responde a los medicamentos. Una opción es la estimulación cerebral profunda (DBS), en la que se implantan electrodos en el cerebro y se conectan a un pequeño dispositivo eléctrico llamado generador de pulsos para estimular sin dolor al cerebro para bloquear las señales que causan muchos de los síntomas motores de la EP. La DBS es generalmente apropiada para personas con EP sensible a levodopa que han desarrollado discinesias u otros síntomas incapacitantes “apagados” a pesar de la terapia con medicamentos. Sin embargo, el DBS no impide que la EP progrese y algunos problemas pueden regresar gradualmente.
Enfermedad de Huntingtons
La enfermedad de Huntington (HD) es un trastorno hereditario que causa la muerte de poblaciones seleccionadas de neuronas. Las neuronas afectadas se localizan en diversas áreas del cerebro, incluyendo aquellas en los ganglios basales que ayudan a controlar el movimiento voluntario (intencional).
Síntomas de la EH
Los síntomas de la enfermedad, que empeora progresivamente, incluyen movimientos incontrolados (corea), posturas corporales anormales y cambios en el comportamiento, la emoción, el juicio y la cognición. Las personas con EH también desarrollan problemas de coordinación, dificultad para hablar y dificultad para alimentarse y tragar.
La EH suele comenzar entre los 30 y los 50 años. Una forma de inicio más temprana llamada EH juvenil ocurre menores de 20 años. Sus síntomas difieren algo de la EH de inicio en adultos e incluyen rigidez, lentitud, dificultad en la escuela, sacudidas musculares involuntarias rápidas llamadas mioclono y convulsiones. Más de 30 mil estadounidenses tienen HD.
La enfermedad de Huntington causa discapacidad que empeora con el tiempo. Actualmente no hay ningún tratamiento disponible para ralentizar, detener o revertir el curso de la EH. Las personas con EH generalmente mueren dentro de los 10 a 30 años después de la aparición de los síntomas, más comúnmente por infecciones (más a menudo neumonía) y lesiones relacionadas con caídas.
Causas de la EH
La enfermedad de Huntington es causada por una mutación en el gen de una proteína llamada huntingtina. El gen mutado incluye un mayor número de repeticiones de una porción selecta de su código genético normal. Todavía no está claro cómo estas repeticiones aumentadas conducen al trastorno. Cada hijo de un padre con EH tiene una probabilidad de 50-50 de heredar el gen mutado. Un niño que no hereda el gen de la EH no desarrollará la enfermedad y no podrá transmitirla a las generaciones posteriores. Dado que este es un rasgo autosómico dominante, una persona que hereda el gen de la EH eventualmente desarrollará la enfermedad. La EH generalmente se diagnostica con base en una prueba genética, antecedentes médicos, imágenes cerebrales y pruebas neurológicas y de laboratorio.
Tratamiento de la EH
No existe ningún tratamiento que pueda detener o revertir el curso de la EH. Los fármacos, tetrabenazina y deuterabenazina pueden tratar la corea asociada a la EH. Los antipsicóticos pueden aliviar la corea y ayudar a controlar alucinaciones, delirios y arrebatos violentos. Se pueden recetar medicamentos para tratar la depresión y la ansiedad. Los efectos secundarios de los medicamentos utilizados para tratar los síntomas de la EH pueden incluir fatiga, sedación, disminución de la concentración, inquietud o hiperexcitabilidad, y solo deben usarse cuando los síntomas crean problemas para el individuo.
Esclerosis lateral amiotrófica (formalmente conocida como Enfermedad de Lou Gehrig)
La esclerosis lateral amiotrófica (ELA) es una rara enfermedad neurológica que afecta principalmente a las neuronas responsables de controlar el movimiento muscular voluntario. Los músculos voluntarios producen movimientos como masticar, caminar y hablar. La enfermedad es progresiva, lo que significa que los síntomas empeoran con el tiempo. Actualmente, no hay cura para la ELA y no hay tratamiento efectivo para detener o revertir la progresión de la enfermedad.
La ELA pertenece a un grupo más amplio de trastornos conocidos como enfermedades de las neuronas motoras, los cuales son causados por el deterioro gradual (degeneración) y la muerte de las neuronas motoras. A medida que las neuronas motoras degeneran, dejan de enviar mensajes a los músculos y los músculos se debilitan gradualmente, comienzan a contraerse y se desperdician (atrofia). Eventualmente, el cerebro pierde su capacidad para iniciar y controlar movimientos voluntarios.
Los primeros síntomas de ELA suelen incluir debilidad muscular o rigidez. Poco a poco todos los músculos voluntarios se ven afectados, y los individuos pierden su fuerza y la capacidad de hablar, comer, moverse e incluso respirar. La mayoría de las personas con ELA mueren por insuficiencia respiratoria, generalmente dentro de los tres a cinco años desde que aparecen los síntomas por primera vez. No obstante, alrededor del 10 por ciento de las personas con ELA sobreviven 10 o más años.
Debido a que las personas con ELA generalmente pueden realizar procesos mentales superiores como el razonamiento, el recuerdo, la comprensión y la resolución de problemas, son conscientes de su pérdida progresiva de función y pueden volverse ansiosos y deprimidos. Un pequeño porcentaje de individuos puede experimentar problemas con el lenguaje o la toma de decisiones, y cada vez hay más evidencia de que algunos incluso pueden desarrollar una forma de demencia con el tiempo.
Posibles causas de ELA
Se desconoce la causa de la ELA, y los científicos aún no saben por qué la ELA golpea a algunas personas y no a otras.
La hiperexcitabilidad neuronal motora contribuye potencialmente a la muerte de las neuronas motoras en ELA. Muchos estudios previos apoyan esta hipótesis, incluyendo ensayos clínicos de riluzol, un antagonista del glutamato que retrasa la progresión de la ELA (Miller et al., 2012).
Además del posible papel de la disfunción de las neuronas motoras, la evidencia científica sugiere que tanto la genética como el ambiente juegan un papel en la degeneración de las neuronas motoras y el desarrollo de ELA.
En 1993, científicos apoyados por el Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares (NINDS) descubrieron que las mutaciones en el gen SOD1 se asociaban con algunos casos de ELA familiar. Desde entonces, se han identificado más de una docena de mutaciones genéticas adicionales, muchas a través de investigaciones apoyadas por NINDS.
La investigación sobre ciertas mutaciones genéticas sugiere que los cambios en el procesamiento de las moléculas de ARN pueden conducir a la degeneración de las neuronas motoras relacionadas con el ALS. Las moléculas de ARN están involucradas con la producción de moléculas en la célula y con la actividad génica.
Otras mutaciones genéticas indican que puede haber defectos en el reciclaje de proteínas, un proceso natural en el que las proteínas que funcionan mal se descomponen y se utilizan para construir nuevas proteínas que funcionan. Otros señalan posibles defectos en la estructura y forma de las neuronas motoras, así como una mayor susceptibilidad a las toxinas ambientales.
Los investigadores están estudiando el impacto de factores ambientales, como la exposición a agentes tóxicos o infecciosos, virus, trauma físico, dieta y factores conductuales y ocupacionales. Por ejemplo, la exposición a toxinas durante la guerra, o la actividad física extenuante, son posibles razones por las que algunos veteranos y atletas pueden estar en mayor riesgo de desarrollar ELA. La investigación en curso puede mostrar que algunos factores están involucrados en el desarrollo o progresión de la enfermedad.
Atribuciones:
Selecciones de secciones de Enfermedad de Parkinson y Esclerosis Múltiple adaptadas de Neuroscience Canadian, 2da Edición. Licencia: CC-BY 4.0.
Esclerosis Lateral Amiotrófica sección adaptada del Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares (NINDS). Licencia: Dominio Público: No Conocido Derechos de Autor