
6.6: Polímeros
- Page ID
- 126190

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Conceptos básicos
Los polímeros de cadena lineal son estructuras repetitivas con la fórmula química\((A)\ns_x\), donde\(A\) está la unidad de fórmula y\(x\) es el grado de polimerización. En muchos casos (poliestireno), no\(x\gtwid 10^5\) es infrecuente. Para una introducción muy legible al tema, véase P. G. de Gennes, Scaling Concepts in Polymer Physics.
Muy a menudo una solución polimérica dada contendrá una distribución de\(x\) valores; esto se conoce como polidispersidad. Diversas técnicas de preparación, como la cromatografía, pueden mitigar el grado de polidispersidad. Otra característica morfológica de los polímeros es la ramificación, en la que los polímeros no forman cadenas lineales.

Los polímeros exhiben una flexibilidad estática que se puede entender como sigue. Considera un hidrocarburo de cadena larga con una\(-\RC-\RC-\RC-\) cadena principal. El ángulo entre\(\RC-\RC\) enlaces sucesivos se fija en\(\theta\approx 68^\circ\), pero el ángulo azimutal\(\vphi\) puede tomar uno de los tres posibles valores de baja energía, como se muestra en el panel derecho de la Figura\(\PageIndex{2}\). Así, las probabilidades relativas de las orientaciones gauche y trans son
\[{{Prob}\,({gauche})\over{Prob}\,({trans})} = 2\,e^{-\RDelta \ve/\kT}\]
donde\(\RDelta \ve\) está la diferencia de energía entre las configuraciones trans y gauche. Esto significa que la cadena de polímero es de hecho una bobina aleatoria con una longitud de persistencia
\[\ell\ns_\Rp = \ell\ns_0 \, e^{\RDelta\ve/\kT}\]
donde\(\ell\ns_0\) es una escala microscópica de longitud, dada aproximadamente por la longitud de una unidad de fórmula, que es aproximadamente de unos pocos Ångstroms (ver Figura\(\PageIndex{2}\)). Dejar\(L\) ser la longitud total del polímero cuando se estira en línea recta. Si\(\ell\ns_\Rp > L\), el polímero es rígido. Si\(\ell\ns_\Rp \ll L\), el polímero es rígido en la escala de longitud\(\ell\ns_\Rp\) pero flexible en escalas más largas. Tenemos
\[{\ell\ns_\Rp\over L}={1\over N}\,e^{\RDelta\ve/\kT}\quad ,\]
donde ahora usamos\(N\) (en lugar de\(x\)) para el grado de polimerización.
En el dominio del tiempo, el polímero presenta una flexibilidad dinámica en escalas mayores que un tiempo de persistencia. El tiempo de persistencia\(\tau\ns_\Rp\) es el tiempo requerido para una transición transgauche. La velocidad para tales transiciones es establecida por la barrera de energía que\(B\) separa las configuraciones trans de gauche:
\[\tau\ns_\Rp=\tau\ns_0\,e^{B/\kT}\]
donde\(\tau\ns_0\sim 10^{-11}\,\Rs\). En escalas de frecuencia\(\omega\ll \tau_\Rp^{-1}\) el polímero es dinámicamente flexible. Si\(\RDelta\ve\sim\kT \ll B\) el polímero es flexible desde un punto de vista estático, pero dinámicamente rígido. Es decir, hay muchas orientaciones gauche de sucesivos enlaces de carbono que reflejan un desorden apagado. El polímero luego forma una bobina aleatoria congelada, como una percha retorcida.

Polímeros como caminatas aleatorias
Un polímero se puede modelar mediante una caminata aleatoria autoevitable (SAW). Es decir, en escalas más largas que\(\ell\ns_\Rp\), se retuerce aproximadamente aleatoriamente en el espacio sujeto a la restricción de que no se superpone a sí misma. Antes de considerar las matemáticas de las sierras, primero recordemos algunos aspectos de las caminatas aleatorias ordinarias que no son autoevitables.
Simplificaremos aún más las cosas considerando caminatas aleatorias en una celosía hipercúbica de dimensión\(d\). Tal celosía tiene número de coordinación\(2d\), hay vectores de separación vecinos\(2d\) más cercanos, dados por\(\Bdelta = \pm a\,\ehat\ns_1 \, , \, \pm a\,\ehat\ns_2 \, , \, \ldots \, , \, \pm a\,\ehat\ns_d\), donde\(a\) está el espaciado de celosía. Considera ahora un paseo aleatorio de\(N\) pasos comenzando por el origen. Después de\(N\) los pasos la posición es
\[\BR\ns_N=\sum_{j=1}^N\Bdelta\ns_j\]
donde\(\Bdelta\ns_j\) toma uno de los valores\(2d\) posibles. Ahora ya no\(N\) es el grado de polimerización, sino algo aproximado\(L/\ell\ns_\Rp\), que es el número de longitudes de persistencia en la cadena. Asumimos que cada paso es independiente, de ahí\(\langle \delta_j^\alpha\,\delta_{j'}^\beta\rangle=(a^2/d)\,\delta\ns_{jj'}\delta^{\alpha\beta}\) y\(\blangle \BR_N^2\brangle=N a^2\). La distribución completa\(P\ns_N(\BR)\) viene dada por
\[\begin{split} P\ns_N(\BR)&=(2d)^{-N}\sum_{\Bdelta\ns_1} \cdots\sum_{\Bdelta\ns_N} \delta\ns_{\BR,\sum_j\Bdelta\ns_j} \\ &= a^d\!\!\! \int\limits_{-\pi/a}^{\pi/a}\! \!{dk\ns_1\over 2\pi} \cdots\!\!\! \int\limits_{-\pi/a}^{\pi/a}\! \!{dk\ns_d\over 2\pi} \>e^{-i\Bk\cdot\BR} \> \Bigg[ {1\over d} \sum_{\mu=1}^d \cos(k\ns_\mu a) \Bigg]^N \\ &=a^d\!\int\limits_{\HOmega}\!\!{d^d\!k\over (2\pi)^d} \> e^{-i\Bk\cdot\BR} \, \exp\!\Bigg[ N\ln\!\bigg( 1 - {1\over 2d} \,\Bk^2 a^2 + \ldots \bigg) \Bigg] \\ &\approx \bigg({a\over 2d}\bigg)^{\!d}\int\!\! d^d\!k \> e^{-N\Bk^2 a^2/2d}\, e^{-i\Bk\cdot\BR} = \bigg({d\over 2\pi N}\bigg)^{\!d/2}\, e^{-d\BR^2/2Na^2}\quad . \label{polydist} \end{split}\]
Se trata de un sencillo gaussiano, con ancho\(\blangle \BR^2 \brangle = d\ncdot(Na^2/d)=Na^2\), como ya hemos calculado. La cantidad\(\BR\) definida aquí es el vector de extremo a extremo de la cadena. La distancia RMS de extremo a extremo es entonces\(\langle\BR^2\rangle^{1/2}=\sqrt{N} a\equiv R\ns_0\).
Una figura relacionada de mérito es el radio de giro,\(R\ns_\Rg\), definido por
\[R_\Rg^2={1\over N}\Big\langle \sum_{n=1}^N\big(\BR\ns_n-\BR\ns_{\ssr{CM}}\big)^2 \Big\rangle \quad,\]
donde\(R\ns_{\ssr{CM}}={1\over N}\sum_{j=1}^N \BR\ns_j\) está la posición del centro de masa. Un breve cálculo rinde
\[R_g^2=\big(N+3-4N^{-1}\big)\,a^2\sim {Na^2\over 6}\quad,\]
en todas las dimensiones.
El número total de configuraciones de caminata aleatoria con vector de extremo a extremo\(\BR\) es entonces\((2d)^N P\ns_N(\BR)\), por lo que la entropía de una cadena en elongación fija es
\[S(\BR,N)=\kB\ln\Big[ (2d)^N P\ns_N(\BR)\Big] = S(0,N) - {d\kB \BR^2\over 2 Na^2}\quad .\]
Si asumimos que la energía de la cadena es independiente de la conformación, entonces\(E=E\ns_0(N)\) y
\[F(\BR,N)=F(0,N) + {d\kT \BR^2\over 2 Na^2}\quad .\]
En presencia de una fuerza externa\(F\ns_{ext}\), la energía libre de Gibbs es la transformación de Legendre
\[G(\BF\ns_{ext},N)=F(\BR,N)-\BF\ns_{ext}\cdot\BR\quad ,\]
y\(\pz G/\pz\BR=0\) luego da la relación
\[\blangle \BR(\BF\ns_{ext},N)\brangle = {Na^2\over d\kT}\,\BF\ns_{ext}\quad .\]
Esto puede considerarse una ecuación de estado para el polímero.
Siguiendo de Gennes, consideremos una cadena con cargas\(\pm e\) en cada extremo, colocada en un campo eléctrico externo de magnitud\(E=30,000\,\RV/{cm}\). Vamos\(N=10^4\),\(a=2\,\) Å, y\(d=3\). ¿Cuál es el alargamiento? De la fórmula anterior, tenemos
\[{R\over R\ns_0} = {eE R\ns_0\over 3\kT} = 0.8 \ ,\]
con\(R\ns_0=\sqrt{N} a\) como antes.

Factor de estructura
También podemos calcular el factor de estructura,
\[ \begin{align*} S(\Bk) &={1\over N}\,\Big\langle\sum_{m=1}^N\sum_{n=1}^N e^{i\Bk\cdot(\BR\ns_m-\BR\ns_n)}\Big\rangle\\[4pt] &=1+{2\over N}\,\sum_{m=1}^N\sum_{n=1}^{m-1} \Big\langle e^{i\Bk\cdot(\BR\ns_m-\BR\ns_n)}\Big\rangle \end{align*}\]
Para promedios con respecto a una distribución gaussiana,
\[\blangle e^{i\Bk\cdot(\BR\ns_m-\BR\ns_n)} \brangle = \exp\bigg\{\!-{1\over 2}\>\Big\langle \big(\Bk\cdot(\BR\ns_m-\BR\ns_n)\big)^2 \Big\rangle\bigg\}\quad .\]
Ahora para\(m>n\) nosotros tenemos\(\BR\ns_m-\BR\ns_n=\sum_{j=n+1}^m \Bdelta\ns_j\), y por lo tanto
\[\Big\langle \big(\Bk\cdot(\BR\ns_m-\BR\ns_n)\big)^2 \Big\rangle=\sum_{j=n+1}^m \blangle (\Bk\cdot \Bdelta\ns_j)^2\brangle = {1\over d}\,(m-n)\,\Bk^2 a^2\quad,\]
ya que\(\langle \delta_j^\alpha\,\delta_{j'}^\beta\rangle=(a^2/d)\,\delta\ns_{jj'}\delta^{\alpha\beta}\). Entonces tenemos
\[S(\Bk)=1+{2\over N}\sum_{m=1}^N\sum_{n=1}^{m-1} e^{-(m-n) \,\Bk^2 a^2/2d}\\ ={N\,(e^{2\mu_\Bk}-1) -2\, e^{\mu_\Bk}\, (1-e^{-N\mu_\Bk})\over N\big(e^{\mu_\Bk}-1\big)^2}\quad ,\]
donde\(\mu_\Bk=\Bk^2 a^2/2d\). En el límite donde\(N\to\infty\) y\(a\to 0\) con\(Na^2=R_0^2\) constante, el factor de estructura tiene una forma de escalado,\(S(\Bk)=N f(N\mu_\Bk)=(R\ns_0/a)\,f(k^2R_0^2/2d)\), donde
\[f(x)={2\over x^2}\,\big(e^{-x}-1+x\big)= 1 - {x\over 3} + {x^2\over 12} + \ldots\ .\]
Modelo Rouse
Considera a continuación una cadena de polímero sometida a forzamiento estocástico. Modelamos la cadena como una colección de puntos de masa conectados por resortes, con una energía potencial\(U=\half k \sum_n \big(\Bx\nd_{n+1}-\Bx\nd_n\big)^2\). Esto reproduce la distribución de la Ecuación\ ref {polydist} si tomamos la constante de resorte para ser\(k=3\kT/a^2\) y establecemos la longitud de equilibrio de cada resorte a cero. Las ecuaciones de movimiento son entonces
\[M{\ddot\Bx}\nd_n + \gamma\,{\dot\Bx}\nd_n=-k\big(2\Bx\nd_n-\Bx\nd_{n-1}-\Bx\nd_{n+1}\big)+\Bf\nd_n(t)\quad,\]
donde\(n\in\{1,\ldots,N\}\) y\(\{f^\mu_n(t)\}\) un conjunto de forzamientos de ruido blanco gaussiano, cada uno con media cero, y
\[\blangle f^\mu_n(t)\,f^\nu_{n'}(t')\brangle=2\gamma\kT\,\delta\nd_{nn'}\,\delta^{\mu\nu}\,\delta(t-t')\quad.\]
Definimos\(\Bx\nd_0\equiv \Bx\nd_1\) y\(\Bx\nd_{N+1}\equiv\Bx\nd_N\) para que la masa final apunte\(n=1\) y\(n=N\) experimente una fuerza restauradora de un solo vecino. Suponemos que la cadena está sobreamortiguada y establecida\(M\to 0\). Entonces tenemos
\[\gamma\,{\dot\Bx}\nd_n=-k\sum_{n'=1}^N A\nd_{nn'}\,\Bx\nd_{n'} + \Bf\nd_n(t)\quad,\]
donde
\[A=\begin{pmatrix} 1 & -1 & 0 & 0 & \cdots & 0 \\ -1 & 2 & -1 & 0 & \cdots & 0 \\ 0 & -1 & 2 & -1 & \cdots & 0 \\ 0 & 0 & -1 & \ddots & \cdots & \vdots \\ \vdots & & \ddots & \ddots & 2 & -1 \\ 0 & \cdots & \cdots & 0 & -1 & 1 \end{pmatrix}\quad.\]
La matriz\(A\) es real y simétrica. Sus funciones propias están etiquetadas\(\psi\nd_j(n)\), con\(j\in\{0,\ldots,N-1\}\):
\[\begin{split} \psi\nd_0(n)&={1\over\sqrt{N}}\\ \psi\nd_j(n)&=\sqrt{2\over N}\,\cos\!\bigg({(2n-1)j\pi\over 2N}\bigg) \quad,\quad j\in\{1,\ldots,N-1\} \end{split}\]
Las relaciones de integridad y ortonormalidad son
\[\sum_{j=0}^{N-1}\psi\nd_j(n)\,\psi\nd_j(n')=\delta\nd_{nn'}\qquad,\qquad\sum_{n=1}^N\psi\nd_j(n)\,\psi\nd_{j'}(n)=\delta\nd_{jj'}\quad,\]
con valores propios\(\lambda\nd_j=4\sin^2\!\big(\pi j/2N\big)\). Tenga en cuenta que\(\lambda\nd_0=0\).
Ahora trabajamos en la base de los modos normales\(\{\eta^\mu_j\}\), donde
\[\eta^\mu_j(t)=\sum_{n=1}^N\psi\nd_j(n)\,x^\mu_n(t)\qquad,\qquad x^\mu_n(t)=\sum_{j=0}^{N-1}\psi\nd_j(n)\,\eta^\mu_j(t)\quad.\]
Entonces tenemos
\[{d\Beta\nd_j\over dt}=-{1\over\tau\nd_j}\,\Beta\nd_j + \Bg\nd_j(t)\quad, \label{RouseNM}\]
donde está el tiempo de\(j^{th}\) relajación
\[\tau\nd_j={\gamma\over 4k\sin^2\!\big(\pi j/2N\big)}\]
y
\[g^\mu_j(t)=\gamma^{-1}\sum_{n=1}^N\psi\nd_j(n)\,f^\mu_n(t)\quad.\]
Tenga en cuenta que
\[\blangle g^\mu_j(t)\,g^\nu_{j'}(t')\brangle=2\gamma^{-1}\kT\,\delta\nd_{jj'}\,\delta^{\mu\nu}\,\delta(t-t')\quad.\]
Integrando Ecuación\ ref {RousenM}, tenemos para,\(j=0\),
\[\Beta\nd_0(t)=\Beta\nd_0(0)+\int\limits_0^t\!\!dt'\>\Bg\nd_0(t')\quad.\]
Para los\(j>0\) modos,
\[\Beta\nd_j(t)=\Beta\nd_j(0)\,e^{-t/\tau\nd_j}+\int\limits_0^t\!\!dt'\>\Bg\nd_j(t')\,e^{(t'-t)/\tau\nd_j}\quad.\]
Por lo tanto,
\[\begin{split} \blangle\eta^\mu_0(t)\,\eta^\nu_0(t')\brangle\nd_\Rc&=2\gamma^{-1}\kT\,\delta^{\mu\nu}\,\textsf{min}(t,t')\\ \blangle\eta^\mu_j(t)\,\eta^\nu_j(t')\brangle\nd_\Rc&=\gamma^{-1}\kT\,\delta^{\mu\nu}\,\tau\nd_j\,\Big( e^{-|t-t'|/\tau\nd_j}-e^{-(t+t')/\tau\nd_j}\Big)\quad, \end{split}\]
donde se define como el 'promedio conectado'\(\langle A(t)\,B(t')\rangle\nd_\Rc\equiv \langle A(t)\,B(t')\rangle-\langle A(t)\rangle\langle B(t')\rangle\). Transformando de nuevo a la base original del espacio real, entonces tenemos
\[\blangle x^\mu_n(t)\,x^\nu_{n'}(t')\brangle\nd_\Rc = {2\kT\over N\gamma }\,\delta^{\mu\nu}\textsf{min}(t,t') + {\kT\over \gamma}\delta^{\mu\nu}\sum_{j=1}^{N-1} \tau\nd_j\,\psi\nd_j(n)\,\psi\nd_{j}(n')\,\Big( e^{-|t-t'|/\tau\nd_j}-e^{-(t+t')/\tau\nd_j}\Big)\quad.\]
En particular, la 'variación conectada' de\(\Bx\nd_n(t)\) es
\[\textsf{CVar}\big[\Bx\ns_n(t)\big]\equiv \blangle \big[\Bx\nd_n(t)\big]^2\brangle\nd_\Rc={6\kT\over N\gamma}\,t + {3\kT\over\gamma}\sum_{j=1}^{N-1} \tau\nd_j\,\big[\psi\nd_j(n)\big]^2\,\big(1-e^{-2t/\tau\nd_j}\big)\quad. \label{Rousevar}\]
De esto vemos que en tiempos largos, cuando\(t\gg\tau\ns_1\), el movimiento de\(\Bx\nd_n(t)\) es difusivo, con constante de difusión\(D=\kT/N\gamma\propto B^{-1}\), que es inversamente proporcional a la longitud de la cadena. Recordemos el resultado de Stokes\(\gamma=6\pi\eta R/M\) para una esfera de radio\(R\) y masa que\(M\) se mueve en un fluido de viscosidad dinámica\(\eta\). De\(D=\kT/\gamma M\), ¿no deberíamos esperar que la constante de difusión sea\(D=\kT/6\pi\eta R\propto N^{-1/2}\), ya que el radio de giro del polímero es\(R\nd_\Rg\propto N^{1/2}\)? Este argumento pasa de contrabando en el supuesto de que la única disipación se está produciendo en la superficie exterior del polímero, modelada como una bola de radio\(R\ns_\Rg\). De hecho, para una caminata aleatoria gaussiana en tres dimensiones espaciales, la densidad para\(r<R\ns_\Rg\) es\(\rho\propto N^{-1/2}\) ya que hay\(N\) monómeros dentro de una región de volumen\(\big(\sqrt{N}\,\big)^3\). Contabilizando la hinchazón de Flory debido a interacciones estéricas (ver más abajo), la densidad es\(\rho\sim N^{-4/5}\), que es aún menor. Así como\(N\to\infty\), la densidad dentro de la esfera\(r=R\ns_\Rg\) efectiva se vuelve pequeña, lo que significa que las moléculas de agua pueden penetrar fácilmente, en cuyo caso toda la cadena polimérica debe considerarse que está en un ambiente disipativo, que es lo que dice el modelo de Rouse: cada monómero ejecutó un movimiento sobreamortiguado.
Un cuidadoso análisis de la Ecuación\ ref {Rousevar} revela que hay un régimen sumiso 12 donde\(\textsf{CVar}\big[\Bx\ns_n(t)\big]\propto t^{1/2}\). Para ver esto, primero toma el\(N\gg 1\) límite, en cuyo caso podemos escribir\(\tau\ns_j=N^2\tau\ns_0/j^2\), dónde\(\tau\ns_0\equiv \gamma/\pi^2 k\) y\(j\in\{1,\ldots,N-1\}\). Dejar\(s\equiv (n-\half)/N \in [0,1]\) ser la coordenada escalada a lo largo de la cadena. El segundo término en la Ecuación\ ref {Rousevar} es entonces
\[S(s,t)\equiv {6\kT\over\gamma}\cdot{\tau\ns_1\over N}\sum_{j=1}^{N-1} {\cos^2(\pi j s)\over j^2}\,\big(1-e^{-2 j^2 t/\tau\ns_1}\big)\quad. \label{RouseS}\]
Vamos\(\sigma\equiv (t/\tau\ns_1)^{1/2}\). Cuando\(t\ll\tau\ns_1\)\(\sigma\ll 1\),, tenemos
\[S(s,t)\simeq {6\kT\over\gamma}\cdot{\tau\ns_1\over N}\>\sigma\!\!\int\limits_0^{N\sigma} \!\!du\>{\cos^2(\pi u s/\sigma)\over u^2}\,\big(1-e^{-2u^2}\big)\quad.\]
Ya que\(s/\sigma\gg 1\), podemos sustituir el término coseno cuadrado por su promedio\(\half\). Si asumimos además\(N\sigma\gg 1\), lo que significa que estamos en el régimen\(1\ll t/\tau\ns_0\ll N^2\), después de realizar la integral obtenemos el resultado
\[S(s,t)={3\kT\over\gamma}\,\sqrt{2\pi\tau\ns_0 t\,}\quad, \label{SSTRouse}\]
siempre que\(s=\CO(1)\) el sitio no\(n\) esté en ninguno de los extremos de la cadena. El resultado en la Ecuación\ ref {SSTrouse} domina el primer término sobre el RHS de la Ecuación\ ReF {Rousevar} desde entonces\(\tau\ns_0\ll t\ll\tau\ns_1\). Este es el régimen sumiso.
Cuando\(t\gg\tau\ns_1=N^2\tau\ns_0\), el exponencial sobre el RHS de la Ecuación\ ref {ROUSES} es despreciable, y si volvemos a aproximarnos\(\cos^2(\pi j s)\simeq\half\), y extendemos el límite superior sobre la suma hasta el infinito, nos encontramos\(S(t)=(3\kT/\gamma)(\tau\ns_1/N)(\pi^2/6)\propto t^0\), que está dominado por el término principal en el RHS de la Ecuación\ ref {Rousevar}. Este es el régimen difusivo, con\(D=\kT/N\gamma\).
Finalmente, cuando\(t\ll\tau\ns_0\), el factor\(1-\exp(-2t/\tau\ns_j)\) puede expandirse a primer orden en\(t\). Entonces se obtiene\(\textsf{CVar}\big[\Bx\ns_n(t)\big]=(6\kT/\gamma)\,t\), que es independiente de la constante de fuerza\(k\). En este régimen, los monómeros no tienen tiempo para responder a la fuerza de sus vecinos, de ahí que cada uno se difunda independientemente. En escalas de tiempo tan cortas, sin embargo, uno debe verificar para asegurarse de que los efectos inerciales pueden ser ignorados, eso\(t\gg M/\gamma\).
Un defecto grave del modelo Rouse es su predicción del tiempo de relajación del\(j=1\) modo,\(\tau\nd_1\propto N^2\). El resultado observado experimentalmente es\(\tau\nd_1\propto N^{3/2}\). Debemos recalcar aquí que el modelo Rouse se aplica a las cadenas ideales. En la teoría de las soluciones poliméricas, un disolvente theta es aquel en el que las bobinas de polímero actúan como cadenas ideales. Una extensión del modelo de Rouse, debido a mi ex colega de UCSD Bruno Zimm, explica las interacciones mediadas hidrodinámicamente entre cualquier par de 'cuentas' a lo largo de la cadena. Específicamente, el modelo Zimm viene dado por
\[{dx^\mu_n\over dt}=\sum_{n'} H^{\mu\nu}(\Bx\nd_n-\Bx\nd_{n'})\Big[k\big(x^\nu_{n'+1}+x^\nu_{n'-1}-2x^\nu_{n'}\big)+f^\nu_{n'}(t)\Big]\quad,\]
donde
\[H^{\mu\nu}(\BR)={1\over 6\pi\eta R}\big(\delta^{\mu\nu}+\HR^\mu\HR^\nu\big)\]
es conocido como el tensor hidrodinámico Oseen (1927) y surge al calcular la velocidad en un fluido en posición\(\BR\) cuando\(\BF=\Bf\,\delta(\Br)\) se aplica una fuerza puntual en el origen. Normalmente se reemplaza\(H(\BR)\) por su promedio sobre la distribución de equilibrio de las configuraciones de polímero. El modelo de Zimm reproduce más correctamente el comportamiento de los polímeros en\(\theta\) -solventes.
Teoría de Flory de los Caminos Autoevitables
Lo que falta en la energía libre de caminar al azar es el efecto de las interacciones estéricas. Un argumento debido a Flory toma en cuenta estas interacciones en un tratamiento medio de campo. Supongamos que tenemos una cadena de radio\(R\). Entonces la densidad promedio de monómeros dentro de la cadena es\(c=N/R^d\). Suponiendo interacciones de corto alcance, entonces deberíamos agregar un término a la energía libre que efectivamente cuente el número de autointersecciones cercanas de la cadena. Este número debe ser aproximadamente\(Nc\). Así, escribimos
\[F(\BR,N)=F\ns_0 + u(T)\,{N^2\over R^d} + \half d \kT\,{R^2\over Na^2}\quad .\]
La interacción efectiva\(u(T)\) es positiva en el caso de un llamado 'buen solvente'.
La energía libre se minimiza cuando
\[0={\pz F\over\pz R}= -{dvN^2\over R^{d+1}} + d\kT\,{R\over Na^2}\quad ,\]
lo que arroja el resultado
\[R\ns_\RF(N)=\bigg({u a^2\over\kT}\bigg)^{\!\!1/(d+2)}N^{3/(d+2)}\propto N^\nu\quad .\]
Así, obtenemos\(\nu=3/(d+2)\). En\(d=1\) esto dice\(\nu=1\), lo cual es exactamente correcto porque un SAW en no\(d=1\) tiene otra opción que seguir yendo en la misma dirección. En\(d=2\), la teoría de Flory predice\(\nu=\frac{3}{4}\), que también es exacta. En\(d=3\), tenemos\(\nu\ns_{d=3}=\frac{3}{5}\), que es extremadamente cercano al valor numérico\(\nu=0.5880\). La teoría de Flory vuelve a ser exacta en la dimensión crítica superior SAW, que es\(d=4\), donde\(\nu=\half\), correspondiente a una caminata aleatoria gaussiana 13. Lo mejor. Media. Campo. Teoría. Siempre.
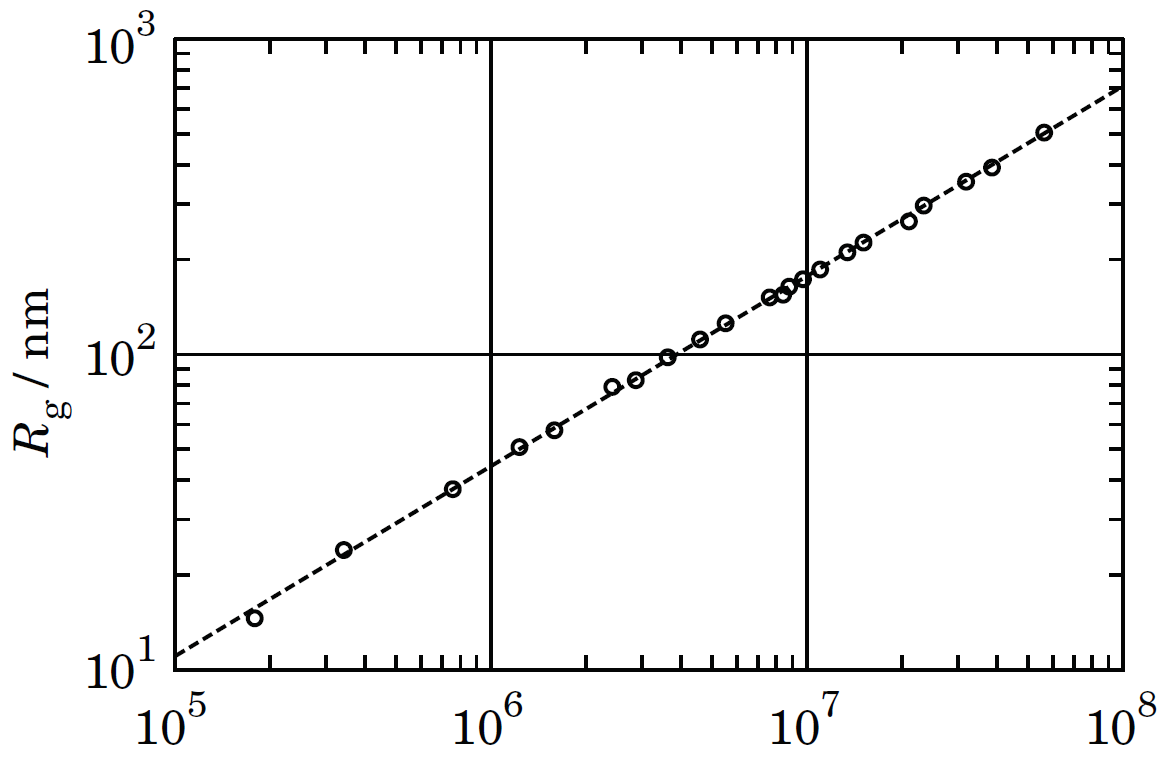
¿Qué tan bien se describen los polímeros como sierras? La figura\(\PageIndex{4}\) muestra el radio de giro\(R\ns_\Rg\) frente al peso molecular\(M\) para cadenas de poliestireno en un disolvente de tolueno y benceno. La pendiente es\(\nu=d\ln R\ns_\Rg/d\ln M=0.5936\). Los resultados experimentales pueden variar con la concentración y la temperatura, pero generalmente confirman la validez del modelo SAW.
Para una SAW bajo una fuerza externa, calculamos la función de partición Gibbs,
\[Y(\BF\ns_{ext},N)=\int \! d^d\!R\>P\ns_N(\BR)\,e^{\BF\ns_{ext}\cdot\BR/\kT} = \int\!d^d\!x\>f(x)\>e^{s\nhat\cdot\Bx}\ ,\]
dónde\(x=R/R\ns_\RF\) y\(s=\kT/R\ns_\RF F\ns_{ext}\) y\(\nhat={\hat\BF}\ns_{ext}\). Uno que tiene\(R(F\ns_{ext})=R\ns_\RF\,\Phi(R\ns_\RF/\xi)\), dónde\(\xi=\kT/F\ns_{ext}\) y\(R(F\ns_{ext})=F\ns_{ext}R^2_\RF/\kT\). Por pequeños valores de su argumento uno tiene\(\Phi(u)\propto u\). Para grandes\(u\) se puede demostrar que\(R(F\ns_{ext})\propto (F\ns_{ext}R\ns_\RF/\kT)^{2/3}\).
En una celosía de número de coordinación\(z\), el número de\(N\) pasos aleatorios -paso a partir del origen es\(\ROmega\ns_N=z^N\). Si limitamos nuestras caminatas aleatorias para que sean autoevitables, el número se reduce a
\[\ROmega^{\ssf{SAW}}_N=\CC\,N^{\gamma-1}\,y^N\quad ,\]
donde\(\CC\) y\(\gamma\) son constantes dependientes de la dimensión, y esperamos\(y \ltwid z-1\), ya que por lo menos un SAW no puede doblarse inmediatamente sobre sí mismo. De hecho, en la celosía cúbica se tiene\(z=6\) pero\(y=4.68\), un poco menos que\(z-1\). Uno encuentra\(\gamma\ns_{d=2}\simeq\frac{4}{3}\) y\(\gamma\ns_{d=3}\simeq \frac{7}{6}\). La distancia RMS de extremo a extremo del SAW es
\[R\ns_\RF=a\,N^\nu\quad ,\]
donde\(a\) y\(\nu\) son constantes\(d\) -dependientes, con\(\nu\ns_{d=1}=1\)\(\nu\ns_{d=2}\simeq\frac{3}{4}\), y\(\nu\ns_{d=3}\simeq\frac{3}{5}\). La distribución\(P\ns_N(\BR)\) tiene una forma de escalado,
\[P\ns_N(R)={1\over R^d_\RF}\,f\bigg({R\over R\ns_\RF}\bigg) \qquad (a \ll R \ll Na)\quad .\]
Uno encuentra
\[f(x)\sim\begin{cases} x^g & x\ll 1 \\ \exp(-x^\delta) & x \gg 1\ , \end{cases}\]
con\(g=(\gamma-1)/\nu\) y\(\delta=1/(1-\nu)\).
Polímeros y Solventes
Considera una solución de polímeros monodispersos de longitud\(N\) en un disolvente. \(\phi\)Sea la concentración adimensional de monómero, así\(\phi/N\) es la concentración adimensional de polímero y\(\phi\ns_\Rs=1-\phi\) es la concentración adimensional de disolvente. (Las concentraciones adimensionales se obtienen dividiendo la concentración dimensionada correspondiente por la densidad general). La entropía de mezcla para tal sistema viene dada por la Ecuación 2.352. Tenemos
\[S\ns_{mix}=-{V\kB\over v\ns_0}\cdot\bigg\{ {1\over N}\phi\ln\phi + (1-\phi)\ln(1-\phi) \bigg\} \ ,\]
donde\(v\ns_0\propto a^3\) es el volumen por monómero. Teniendo en cuenta una interacción entre el monómero y el disolvente, tenemos que la energía libre de la mezcla es
\[{v\ns_0\,F_{mix}\over V\kT}={1\over N}\,\phi\ln\phi+(1-\phi)\ln(1-\phi)+\xhi\,\phi(1-\phi)\ .\]
donde\(\xhi\) está la interacción adimensional polímero-disolvente, llamada parámetro Flory. Esto proporciona una teoría de campo promedio del sistema polímero-disolvente.
La presión osmótica\(\Pi\) se define por
\[\Pi=-{\pz F\ns_{mix}\over \pz V}\bigg|_{N\ns_\Rp} \ ,\]
que es la variación de la energía libre de mezcla con respecto al volumen que mantiene constante el número de polímeros. La concentración de monómero es\(\phi=N N\ns_\Rp v\ns_0/V\), entonces
\[{\pz\over\pz V}\bigg|_{N_\Rp}=-{\phi^2\over N N_\Rp\, v\ns_0}\,{\pz\over\pz\phi}\bigg|_{N_\Rp}\ .\]
Ahora tenemos
\[F_{mix}=N N_\Rp\, \kT\left\{ {1\over N}\,\ln\phi+(\phi^{-1}-1)\ln(1-\phi) +\xhi\,(1-\phi)\right\}\ ,\]
y por lo tanto
\[\Pi ={\kT\over v\ns_0}\Big[(N^{-1}-1)\,\phi-\ln(1-\phi)-\xhi\,\phi^2\Big]\ .\]
En el límite de la concentración de monómero que desaparece\(\phi\to 0\), recuperamos
\[\Pi={\phi\,\kT\over N v\ns_0}\ ,\]
que es la ley de gas ideal para polímeros.
Para\(N^{-1}\ll\phi\ll 1\), ampliamos el logaritmo y obtenemos
\[\begin{split} {v\ns_0\,\Pi\over\kT}&={1\over N}\phi + \half(1-2\xhi)\,\phi^2+\CO(\phi^3)\\ &\approx\half(1-2\xhi)\,\phi^2\ . \label{osmopoly} \end{split}\]
Tenga en cuenta que\(\Pi>0\) sólo si\(\xhi<\half\), cual es la condición para un 'buen solvente'.
De hecho, la Ecuación\ ref {osmopoly} es sólo cualitativamente correcta. En el límite donde\(\xhi\ll\half\), Flory mostró que las bobinas de polímero individuales se comportan tanto como esferas duras de radio\(R\ns_\RF\). La presión osmótica satisface entonces algo análogo a una ecuación de estado virial:
\[\begin{split} {\Pi\over\kT}&={\phi\over Nv\ns_0} + A\,\bigg({\phi\over N v\ns_0}\bigg)^{\!\!2}R_\RF^3 + \ldots \\ &={\phi\over N v\ns_0}\,h(\phi/\phi^*)\ . \end{split}\]
Esto se generaliza a una forma de escalado en la segunda línea, donde\(h(x)\) es una función de escalado, y\(\phi^*=Nv\ns_0/R_\RF^3\propto N^{-4/5}\), asumiendo\(d=3\) y\(\nu=\frac{3}{5}\) a partir de la teoría de Flory. Como\(x=\phi/\phi^*\to 0\), debemos recuperar la ley de gas ideal, así\(h(x)=1+\CO(x)\) en este límite. Para\(x\to\infty\), requerimos que el resultado sea independiente del grado de polimerización\(N\). Esto significa\(h(x)\propto x^p\) con\(\frac{4}{5} p = 1\),\(p=\frac{5}{4}\). El resultado se conoce como la ley des Cloiseaux:
\[{v\ns_0\Pi\over\kT}=C\,\phi^{9/4}\ ,\]
donde\(C\) es una constante. Esto es válido para lo que se conoce como soluciones semi-diluidas, donde\(\phi^*\ll\phi\ll 1\). En el límite denso\(\phi\sim 1\), los resultados no exhiben esta universalidad, y debemos apelar a la teoría del estado líquido, que no es nada divertida.