
4.1: Transiciones de fase de primer orden
- Page ID
- 130046

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Por nuestra experiencia diaria, digamos con hielo de agua, agua líquida y vapor de agua, sabemos que una sustancia química (es decir, un conjunto de muchas partículas similares) puede existir en diferentes estados estables: fases. Una sustancia típica puede tener:
- una fase sólida densa, en la que las fuerzas interatómicas mantienen todos los átomos/moléculas en posiciones relativas prácticamente fijas, con solo pequeñas fluctuaciones térmicas alrededor de ellos;
- una fase líquida, de densidad comparable, en la que las distancias relativas entre átomos o moléculas son casi constantes, pero las partículas son prácticamente libres de moverse unas alrededor de otras, y
- una fase gaseosa, típicamente de una densidad mucho menor, en la que las moléculas son virtualmente libres para moverse alrededor del volumen que contiene. 1
La experiencia también nos dice que en ciertas condiciones, dos fases diferentes pueden estar en equilibrio térmico y químico, digamos, hielo flotando en el agua con la temperatura del punto de congelación. En realidad, en la Sec. 3.4 ya discutimos una teoría cualitativa de uno de esos equilibrios: la coexistencia del condensado de Bose-Einstein con el “vapor” no condensado de partículas similares. Sin embargo, este es un caso bastante excepcional cuando la coexistencia de fases se debe a la naturaleza cuántica de las partículas (bosones) que pueden no interactuar directamente. Con mucha más frecuencia, la formación de diferentes fases, y las transiciones entre ellas, se deben a interacciones repulsivas y atractivas de las partículas, discutidas brevemente en la Sec. 3.5.
Las transiciones de fase a veces se clasifican por su orden. 2 Comenzaré su discusión con las llamadas transiciones de fase de primer orden que cuentan con calor\(\Lambda\) latente distinto de cero, la cantidad de calor que es necesaria para convertir una fase en otra fase completamente, incluso si la temperatura y la presión se mantienen constantes. 3 Desafortunadamente, incluso los modelos “microscópicos” más simples de interacción de partículas, como los discutidos en la Sec. 3.5, dan ecuaciones de estado bastante complejas. (Como recordatorio, incluso el modelo de hardball más simple conduce a la serie (\(3.5.14\)), cuyos coeficientes viriales más altos desafían el cálculo analítico). Es por ello que seguiré la tradición para discutir las transiciones de fase de primer orden utilizando un sencillo modelo fenomenológico sugerido en 1873 por Johannes Diderik van der Waals.
Para su introducción, es útil recordar que en la Sec. 3.5 hemos derivado la Ecuación (\(3.5.13\)) —la ecuación de estado para un gas clásico de partículas que interactúan débilmente, que toma en cuenta (aunque aproximadamente) ambos componentes de interacción necesarios para una descripción realista de la condensación de gas/ licuefacción: la atracción de largo alcance de las partículas y su repulsión de corto alcance. Vamos a reescribir ese resultado de la siguiente manera:
\[P+a\frac{N^2}{V^2} = \frac{NT}{V} \left(1+\frac{Nb}{V}\right).\label{1}\]
Como vimos en la derivación de esta fórmula, el significado físico de la constante\(b\) es el volumen efectivo de espacio tomado por una colisión de pares de partículas — ver Ecuación (\(3.5.10\)). La relación (\ ref {1}) es cuantitativamente válida solo si el segundo término entre paréntesis es pequeño, es decir\(Nb << V\), si el volumen total excluido del movimiento libre de las partículas por sus colisiones es mucho menor que el volumen completo\(V\). Para describir la fase condensada (que llamaré “líquida” 4), necesitamos generalizar esta relación con el caso\(Nb \sim V\). Dado que el volumen efectivo que queda para el movimiento de las partículas es\(V – Nb\), es muy natural hacer el siguiente reemplazo:\(V \rightarrow V – Nb\), en la ecuación de estado del gas ideal. Si también mantenemos en el lado izquierdo el término\((aN^2/V^2\), que describe la atracción de largo alcance de las partículas, obtenemos la ecuación de estado de van der Waals:
Ecuación de Van der Waals:
\[\boxed{P+a\frac{N^2}{V^2} = \frac{NT}{V - Nb} .} \label{2}\]
Una ventaja de este modelo simple es que en el límite de gases raros,\(Nb << V\), vuelve a reducirse a la Ecuación microscópicamente justificada (\ ref {1}). (Para verificar esto, es suficiente con Taylor-expandir el lado derecho de la Ecuación (\ ref {2}) en pequeño\(Nb/V << 1\), y retener solo dos términos iniciales.) Exploremos las propiedades básicas de este modelo.
Frecuentemente es conveniente discutir cualquier ecuación de estado en términos de sus isotermas, es decir, las\(P(V)\) curvas trazadas a constante\(T\). Como muestra la Ecuación (\ ref {2}), en el modelo de van der Waals dicha gráfica depende de cuatro parámetros:\(a\)\(b\),\(N\),, y\(T\), complicando el análisis general del modelo. Para simplificar la tarea, es conveniente introducir variables adimensionales: presión\(p \equiv P/P_c\), volumen\(v \equiv V/V_c\) y temperatura\(t \equiv T/T_c\), normalizadas a sus llamados valores críticos,
\[P_c \equiv \frac{1}{27} \frac{a}{b^2}, \quad V_c \equiv 3Nb, \quad T_c \equiv \frac{8}{27}\frac{a}{b}, \label{3}\]
cuyo significado quedará claro en un minuto. En esta notación, la Ecuación (\ ref {2}) adquiere la siguiente forma,
\[p+\frac{3}{v^2}=\frac{8t}{3v-1},\label{4}\]
de manera que las isotermas normalizadas\(p(v)\) dependan de un solo parámetro, la temperatura normalizada\(t\) — ver Figura\(\PageIndex{1}\).
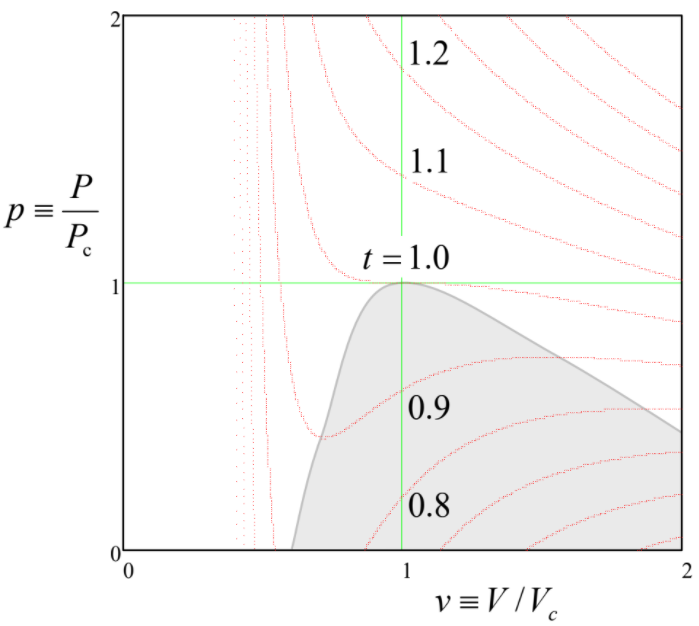
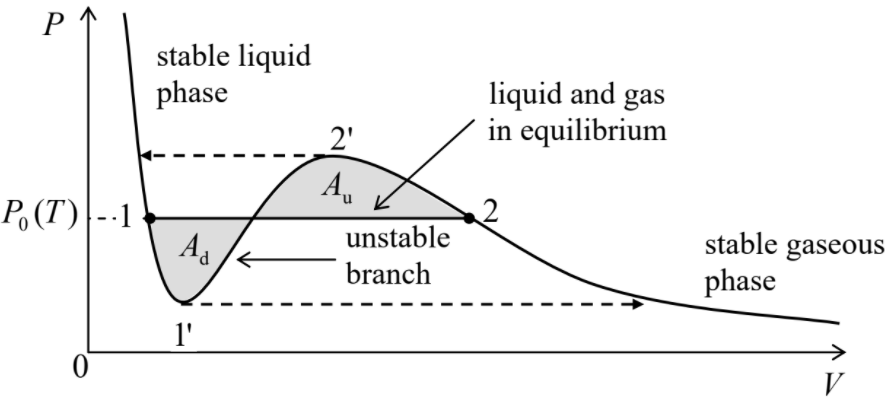
La propiedad más importante de estas parcelas es que las isotermas tienen formas cualitativamente diferentes en dos regiones de temperatura. En\(t > 1\), es decir\(T > T_c\), la presión aumenta monótonamente a la compresión del gas (cualitativamente, como en un gas clásico ideal\(P = NT/V\), con, al que tiende el sistema van der Waals\(T >> T_c\)), es decir, con (\(\partial P/\partial V)_T < 0\)en todos los puntos de la isoterma. 5 Sin embargo, por debajo de la temperatura crítica\(T_c\), cualquier isoterma presenta un segmento con (\(\partial P/\partial V)_T >0\). Es fácil entender que, como mínimo en un experimento de presión constante (ver, por ejemplo, la Figura\(1.4.1\)), 6 estos segmentos describen un equilibrio mecánicamente inestable. En efecto, si debido a una fluctuación aleatoria, el volumen se desvía hacia arriba del valor de equilibrio, la presión también aumentaría, obligando al ambiente (digamos, el pistón pesado en la Figura\(1.4.1\)) a permitir una mayor expansión del sistema, conduciendo a una presión aún mayor, etc. Una desviación similar del volumen a la baja conduciría a una disminución similar al de avalancha del volumen. Dicha inestabilidad de avalancha se desarrollaría cada vez más hasta que el sistema haya alcanzado una de las ramas estables con pendiente negativa\((\partial P/\partial V)_T\). En el rango donde el estado de equilibrio monofásico es inestable, el sistema en su conjunto puede ser estable solo si consta de las dos fases (una con una menor, y otra con una densidad mayor\(n = N/V\)) que son descritas por las dos ramas estables — ver Figura\(\PageIndex{2}\).
Para comprender las propiedades básicas de este sistema bifásico, recordemos las condiciones generales del equilibrio termodinámico de dos sistemas, las cuales han sido discutidas en el Capítulo 1:
Condiciones de equilibrio de fase:
\[\boxed{ T_1 = T_2 \text{ (thermal equilibrium)}, }\label{5}\]
Condiciones de equilibrio de fase:
\[\boxed{\mu_1 = \mu_2 \text{ (“chemical” equilibrium)}, }\label{6}\]
esta última condición significa que la energía promedio de una sola partícula (“sonda”) en ambos sistemas tiene que ser la misma. A esos, hay que añadir la condición evidente del equilibrio mecánico,
Condiciones de equilibrio de fase:
\[\boxed{ P_1 = P_2 \text{ (mechanical equilibrium)}, } \label{7}\]
que se desprende inmediatamente del equilibrio de fuerzas normales ejercidas sobre una frontera entre fases.
Si discutimos isotermas, la Ecuación (\ ref {5}) se cumple automáticamente, mientras que Ecuación (\ ref {7}) significa que la isoterma efectiva que\(P(V)\) describe un sistema bifásico debe ser una línea horizontal — ver Figura\(\PageIndex{2}\):
\[P=P_0 (T). \label{8}\]
A lo largo de esta línea, no cambian 7 propiedades internas de cada fase; solo lo es la distribución de partículas: evoluciona gradualmente desde todas las partículas que están en fase líquida en el punto 1 hasta todas las partículas que están en la fase gaseosa en el punto 2. 8 En particular, según la Ecuación (\ ref {6}), los potenciales químicos\(\mu\) de las fases deben ser iguales en cada punto de la línea horizontal (\ ref {8}). Este hecho nos permite encontrar la posición de la línea: tiene que conectar los puntos 1 y 2 en que los potenciales químicos de las dos fases son iguales entre sí. Reformulemos esta condición como
\[\int^2_1d\mu =0, \quad \text{ i.e. } \int^2_1 dG = 0, \label{9}\]
donde la integral puede tomarse a lo largo de la isoterma monofásica. (Para este cálculo matemático, la inestabilidad mecánica de los estados en alguna parte de esta curva no es importante.) Por su construcción, a lo largo de esa curva,\(N =\)\(T =\) const y const, de manera que según la ecuación (\(1.5.4\))\(dG = –SdT + VdP +\mu dN\),, para un cambio lento (reversible),\(dG = VdP\). De ahí que la ecuación (\ ref {9}) rinde
\[\int^2_1 VdP=0.\label{10}\]
Esta igualdad significa que en Figura\(\PageIndex{2}\), las áreas sombreadas\(A_d\) y\(A_u\) deben ser iguales. 9
Como muestra la misma\(\PageIndex{2}\) figura de la figura, la regla Maxwell puede ser reescrita en una forma diferente,
Regla de área igual de Maxwell:
\[\boxed{\int^2_1 [P-P_0(T)]dV=0.} \label{11}\]
lo cual es más conveniente para los cálculos analíticos que la Ecuación (\ ref {10}) si la ecuación de estado puede resolverse explícitamente para\(P\) — como está en el modelo de van der Waals (\ ref {2}). Dicho cálculo (dejado para el ejercicio del lector) muestra que para ese modelo, la dependencia de la temperatura de la presión de vapor saturado a baja\(T\) es exponencial, 10
\[P_{0}(T) \propto P_{\mathrm{c}} \exp \left\{-\frac{\Delta}{T}\right\}, \quad \text { with } \Delta=\frac{a}{b} \equiv \frac{27}{8} T_{\mathrm{c}}, \quad \text { for } T<<T_{\mathrm{c}},\label{12}\]
que corresponde muy bien a la imagen física de la activación térmica de la partícula desde un pozo potencial de profundidad\(\Delta \).
El parámetro de firma de una transición de fase de primer orden, el calor latente de evaporación
Calor latente: definición
\[\boxed{ \Lambda \equiv \int^2_1 dQ,} \label{13}\]
también se puede encontrar por una integración similar a lo largo de la isoterma monofásica. De hecho, usando Equation (\(1.3.6\))\(dQ = TdS\), obtenemos
\[\Lambda = \int^2_1 TdS = T(S_2-S_1). \label{14}\]
Expresemos el lado derecho de la Ecuación (\ ref {14}) a través de la ecuación de estado. Para ello, tomemos la derivada completa de ambos lados de la Ecuación (\ ref {6}) sobre la temperatura, considerando el valor de\(G = N\mu\) para cada fase en función de\(P\) y\(T\), y teniendo en cuenta que según la Ecuación (\ ref {7}),\(P_1 = P_2 = P_0(T)\):
\[\left(\frac{\partial G_{1}}{\partial T}\right)_{P}+\left(\frac{\partial G_{1}}{\partial P}\right)_{T} \frac{d P_{0}}{d T}=\left(\frac{\partial G_{2}}{\partial T}\right)_{P}+\left(\frac{\partial G_{2}}{\partial P}\right)_{T} \frac{d P_{0}}{d T}. \label{15}\]
Según el primero de Eqs. (\(1.4.16\)), la derivada parcial\((\partial G/\partial T)_P\) es apenas menos la entropía, mientras que según la segunda de esas igualdades,\((\partial G/\partial P)_T\) es el volumen. Así, la ecuación (\ ref {15}) se convierte
\[-S_1+V_1 \frac{dP_0}{dT} = -S_2 + V_2 \frac{dP_0}{dT}. \label{16}\]
Resolviendo esta ecuación para\((S_2 – S_1)\), y conectando el resultado a la Ecuación (\ ref {14}), obtenemos la siguiente fórmula de Clapeyron-Clausius:
Fórmula Clapeyron-Clausius:
\[\boxed{ \Lambda = T(V_2-V_1) \frac{dP_0}{dT}.} \label{17}\]
Para el modelo van der Waals, esta fórmula puede ser fácilmente utilizada para el cálculo analítico de\(\Lambda\) en dos límites:\(T << T_c\) y\((T_c – T) << T_c\) — los ejercicios dejados para el lector. En este último límite\(\Lambda \propto (T_c – T)^{1/2}\),, desapareciendo naturalmente a la temperatura crítica.
Finalmente, algunas propiedades importantes del modelo de van der Waals pueden revelarse más fácilmente al observar el conjunto de sus isócoras\(P = P(T)\) para\(V =\) const, en lugar de las isotermas. En efecto, como muestra la Ecuación (\ ref {2}), todas las isócoras monofásicas son líneas rectas. Sin embargo, si interrumpimos estas líneas en los puntos en que la fase única se vuelve metaestable, y las complementamos con la (¡muy no lineal!) dependencia\(P_0(T)\), obtenemos el patrón (llamado diagrama de fases) que se muestra esquemáticamente en la Figura\(\PageIndex{3a}\).

Así, en el modelo de van der Waals, pueden coexistir dos fases, aunque sólo en ciertas condiciones —en particular ,—\(T < T_c\). Ahora una pregunta natural, más general es si es posible la coexistencia de más de dos fases de una misma sustancia. Por ejemplo, ¿pueden el hielo de agua, el agua líquida y el vapor de agua (vapor) estar todos en equilibrio termodinámico? La respuesta está dada esencialmente por la Ecuación (\ ref {6}). A partir de la termodinámica, sabemos que para un sistema uniforme (es decir, una sola fase), la presión y la temperatura definen completamente el potencial químico\(\mu (P, T)\). De ahí que al tratar dos fases, tuvimos que satisfacer solo una condición de equilibrio químico (\ ref {6}) para dos argumentos comunes\(P\) y\(T\). Evidentemente, esto nos deja con un grado extra de libertad, de manera que el equilibrio bifásico es posible dentro de un cierto rango de\(P\) a fijo\(T\) (o viceversa) — ver de nuevo la línea horizontal en la Figura\(\PageIndex{2}\) y la línea en negrita en la Figura\(\PageIndex{3a}\). Ahora bien, si queremos que tres fases estén en equilibrio, necesitamos satisfacer dos ecuaciones para estas variables:
\[\mu_1 (P,T) = \mu_2(P,T)=\mu_3(P,T). \label{18}\]
Normalmente, las funciones\(\mu (P, T)\) son monótonas, de manera que las dos ecuaciones (\ ref {18}) tienen una sola solución, el llamado punto triple\(\{P_t, T_t\}\). Por supuesto, el triple punto\(\{P_t, T_t\}\) de equilibrio entre tres fases no debe confundirse con los puntos críticos\(\{P_c, T_c\}\) de las transiciones entre cada uno de los pares de dos fases. La figura\(\PageIndex{3b}\) muestra, de manera muy esquemática, su relación para un sistema típico de tres fases sólido-líquido-gas. Por ejemplo, el agua, el hielo y el vapor de agua se encuentran en equilibrio en un punto triple correspondiente a\(P_t \approx 0.612\) kPa\(^{13}\) y\(T_t = 273.16\) K. La importancia práctica de este punto de temperatura particular es que por un acuerdo internacional ha sido aceptado para la definición no solo del Kelvin escala de temperatura, pero también de la referencia de la escala Celsius, como 0.01\(^{\circ}\) C, de manera que la temperatura absoluta cero corresponde exactamente a —273.15\(^{\circ}\) C. 14 Más generalmente, puntos triples de sustancias simples purificadas (como\(\ce{H2}\),\(\ce{N2}\),\(\ce{O2}\),, Ar, Hg, y \(\ce{H2O}\)) se utilizan ampliamente para la calibración del termómetro, definiendo las llamadas escalas de temperatura internacionales incluyendo la escala actualmente aceptada ITS-90.
Este análisis puede generalizarse fácilmente a sistemas multicomponentes que consisten en partículas de varios (digamos,\(L\)) tipos. 15 Si dicho sistema mixto está en una sola fase, es decir, es macroscópicamente uniforme, su potencial químico puede definirse por una generalización natural de la Ecuación (\(1.5.4\)):
\[dG = -SdT+VdP+\sum^L_{l=1} \mu^{(l)}dN^{(l)}.\label{19}\]
El último término refleja el hecho de que generalmente, cada fase única no es una sustancia química pura, sino que tiene ciertas concentraciones de todos los demás componentes, por lo que\(\mu^{ (l)}\) puede depender no sólo de\(P\) y\(T\) sino también de las concentraciones\(c^{(l)} \equiv N^{(l)}/N\) de partículas de cada tipo. Si el número total\(N\) de partículas es fijo, el número de concentraciones independientes es\((L – 1)\). Para el equilibrio químico de\(R\) las fases, todos\(R\) los valores de\(\mu_r^{(l)} (r = 1, 2, ..., R)\) tienen que ser iguales para partículas de cada tipo:\(\mu_1^{(l)} = \mu_2^{(l)} = ... = \mu_R^{(l)}\), con cada una\(\mu_r^{(l)}\) dependiendo de\((L – 1)\) las concentraciones\(c_r^{(l)}\), y también sobre\(P\) y\(T\). Este requisito da\(L(R – 1)\) ecuaciones para\((L –1)R\) concentraciones\(c_r^{(l)}\), más dos argumentos comunes\(P\) y\(T\), es decir, para variables\([(L –1)R + 2]\) independientes. Esto significa que el número de fases tiene que satisfacer la limitación
Regla de fase Gibbs:
\[\boxed{ L(R-1) \leq (L-1)R+2, \quad \text{ i.e. } R \leq L +2, } \label{20}\]
donde se puede alcanzar el signo de igualdad en un solo punto en todo el espacio de parámetros. Esta es la regla de la fase Gibbs. Como comprobación de cordura, para un sistema de un solo componente,\(L = 1\), la regla rinde\(R \leq 3\) —exactamente el resultado que ya hemos comentado.