
14.4: El fosfato piroxidal - un cofactor sumidero de electrones
- Page ID
- 2422

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In section 6.5, we were introduced to the concept of coenzymes - small organic molecules that assist enzymes in doing a particular chemical job. We have seen many specific examples since then: SAM as a methyl group donor, ATP as a phosphoryl group donor, Coenzyme A as an activator for acyl transfer reactions, and most recently glutathione as a 'nucleophile for hire' in the cis/trans isomerization described in section 14.2A.
In this section and the next, we will consider two very important coenzymes that help to stabilize carbanion intermediates in many diverse enzymatic reactions. They may be familiar to you already if you occasionally look at the nutritional information on food packages: thiamine diphosphate (TPP) is the diphosphate ester of thiamine (otherwise known as vitamin B1), and pyridoxal phosphate (PLP), is a derivative of vitamin B6.

Unlike plants and many bacteria, humans do not have the biosynthetic enzymes to make these molecules, so we must get them in our diet.
Many enzymes involved in the metabolism of amino acids make use of PLP as a coenzyme. The basic function of PLP is to act as an 'electron sink', the meaning of which will become clear as we examine in detail the mechanisms of some PLP-dependant reactions. Most of these reactions will be familiar - they include amino acid racemizations, eliminations, additions, and retro-aldol or retro-Claisen cleavages. What will be new is the use of PLP to stabilize the carbanion intermediates in each of these reactions.
14.4A: PLP and the Schiff base linkage to lysine
In the first step of virtually all PLP-dependant reactions, the aldehyde group of the coenzyme forms an imine (Schiff base) linkage with a lysine side chain on the enzyme. (In order to help you to focus on the chemistry happening with the amino acid substrates, the PLP molecule in subsequent figures is colored green - it will be very helpful to refer to color figures).
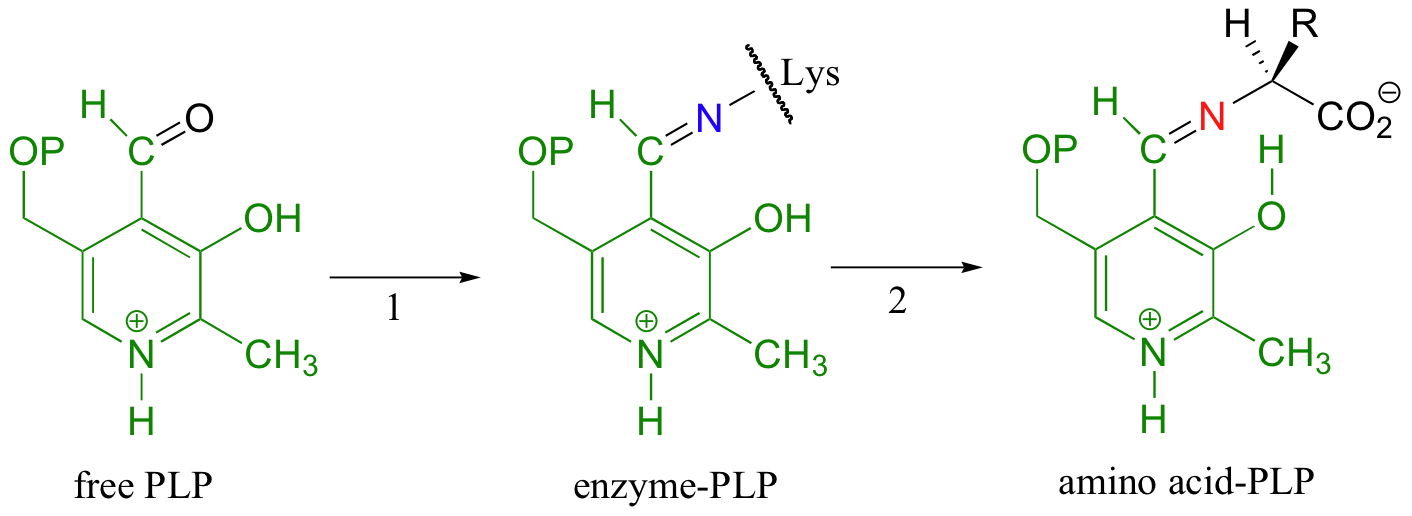
Generally, the next step (step 2 in the figure above) is an imine exchange, as the amine nitrogen on the amino acid substrate replaces the enzyme lysine nitrogen in the imine linkage. This substrate-coenzyme adduct is stabilized by a favorable hydrogen bond between the phenol of PLP and the imine nitrogen.
14.4B: PLP-dependent amino acid racemases
With these preliminaries accomplished, the real PLP chemistry is ready to start. Let's look first at the reaction catalyzed by PLP-dependent alanine racemase. In section 13.2B we saw an example of a PLP-independent amino acid racemase reaction, in which the negatively-charged intermediate was simply the enolate form of a carboxylate.
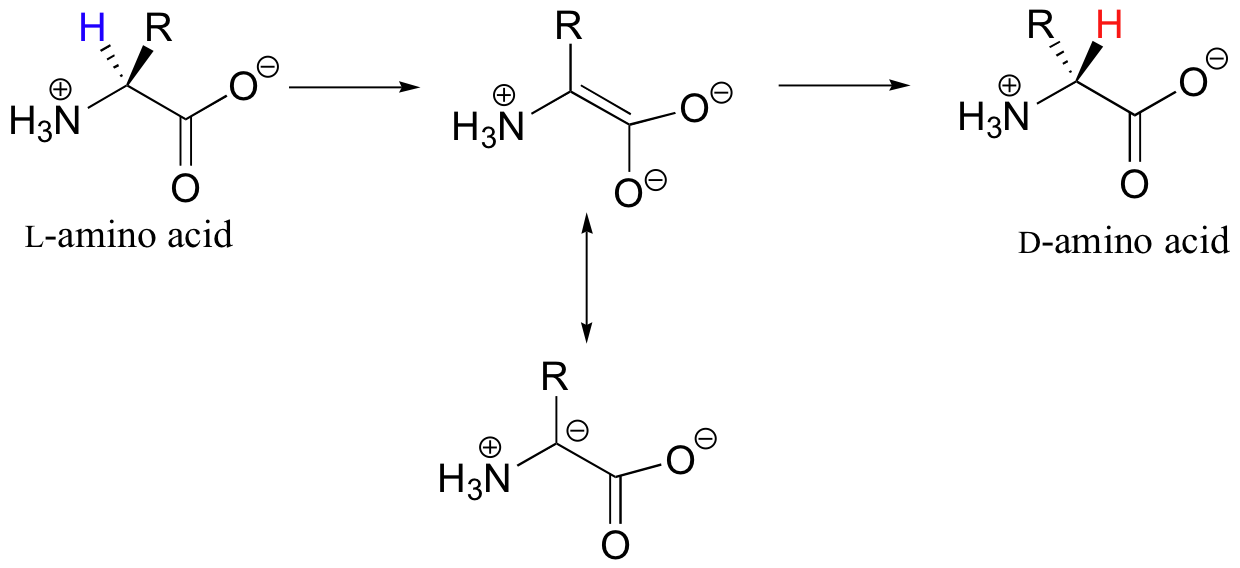
Many other amino acid racemases, however, require the participation of PLP. Once it is linked to PLP in the active site, the a-proton of alanine can be abstracted by an active site base (step 3 below). The negative charge on the carbanion intermediate can, of course, be delocalized to the carboxylate group of alanine. The PLP coenzyme, however, provides a much expanded network of conjugated π-bonds over which the electron density can be delocalized all the way down to the PLP nitrogen.
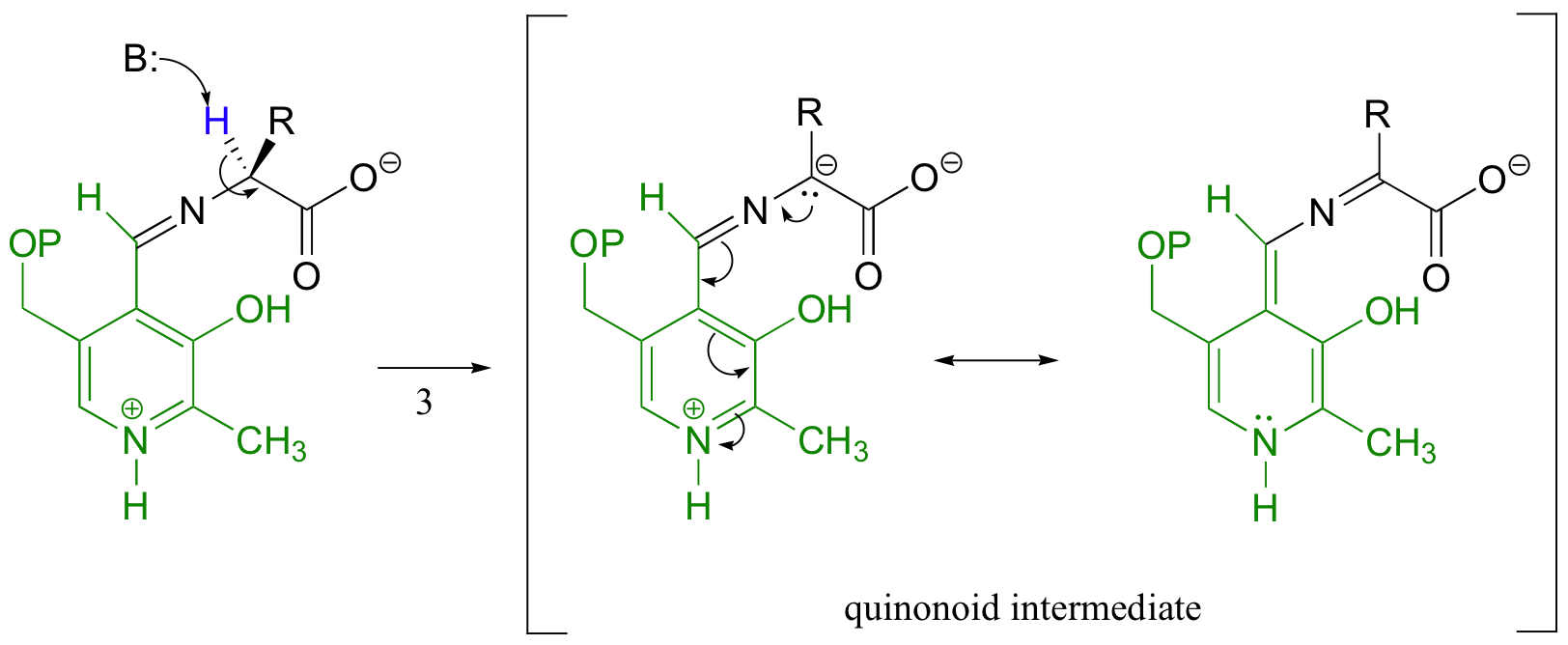
This is what we mean when we say that the job of PLP is to act as an ‘electron sink’: the coenzyme is extremely efficient at absorbing, or delocalizing, the excess electron density on the deprotonated a-carbon of the reaction intermediate. PLP is helping the enzyme to increase the acidity of the a-hydrogen by stabilizing the conjugate base. A PLP-stabilized carbanion intermediate is commonly referred to as a quinonoid intermediate.
In the remaining steps of the alanine racemase reaction, reprotonation occurs on the opposite side of the substrate (step 4), leading to the D-amino acid.
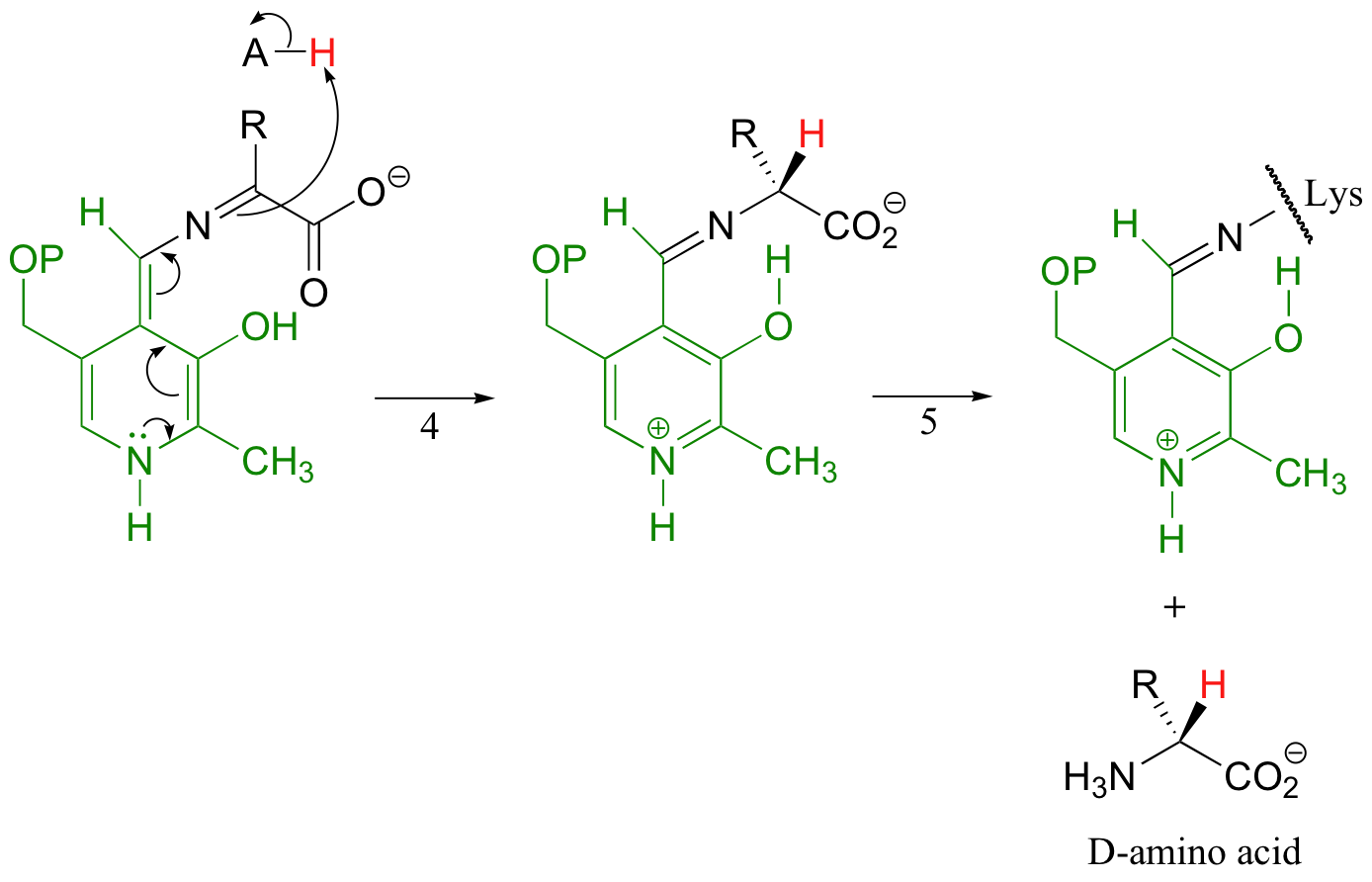
All that remains is another imine exchange (step 5), which frees the D-alanine product and re-attaches PLP to the enzymatic lysine side-chain.
14.4C: PLP-dependent decarboxylation
In the alanine racemase reaction above, PLP assisted in breaking the Cα-H bond of the amino acid. Some PLP-dependant enzymes can catalyze the breaking of the bond between Cα and the carboxylate carbon in an amino acid by stabilizing the resulting carbanion intermediate: these are simply decarboxylation reactions. One of the final steps in the synthesis of lysine is a PLP-dependent decarboxylation.
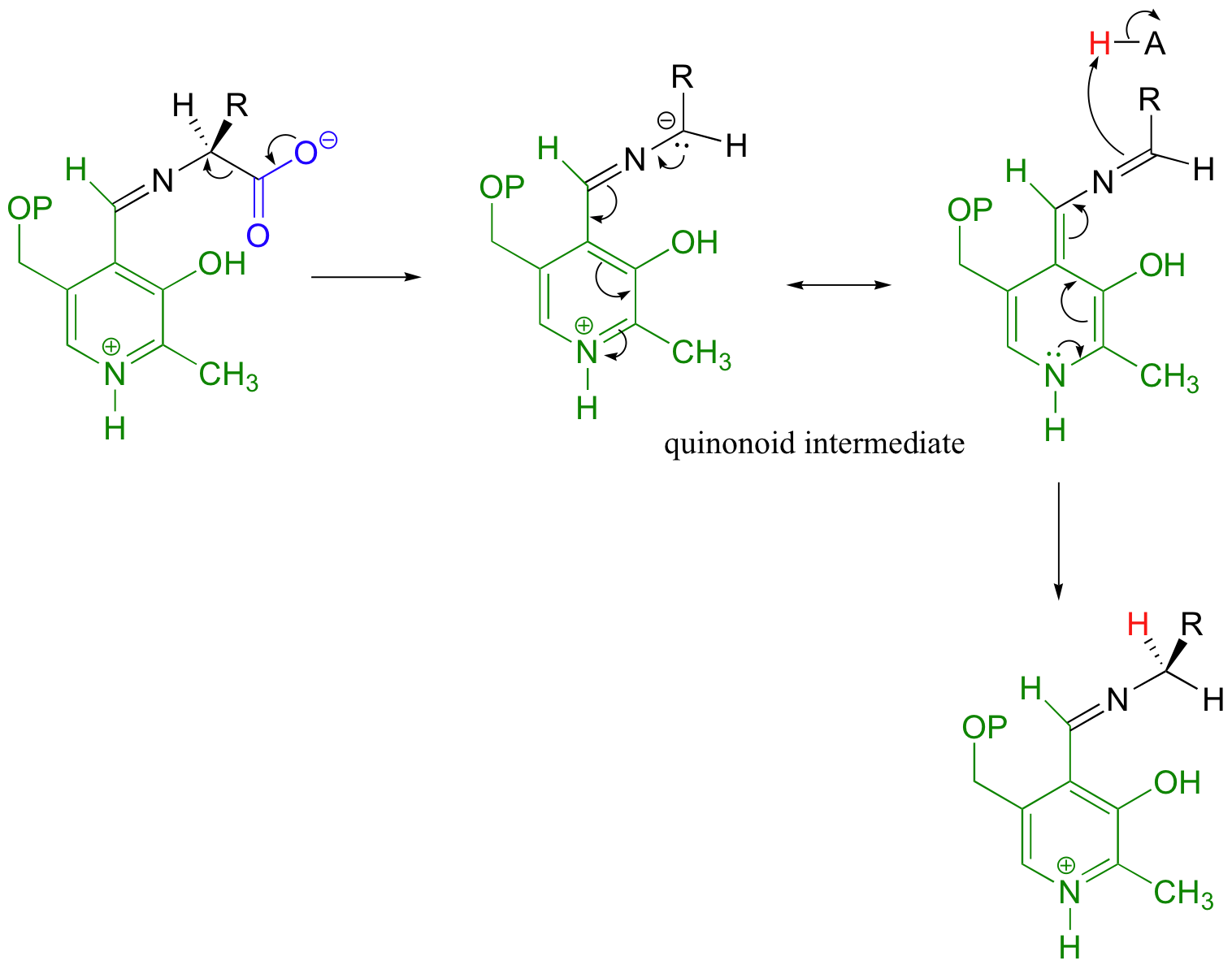
14.4D: PLP-dependent retro-aldol and retro-Claisen reactions
Still other PLP-dependant enzymes catalyze the breaking of the bond between the alpha-carbon and the first carbon on the amino acid side chain - this is referred to as a Cα-Cβ cleavage, and it actually is a variation on the retro-aldol reaction (section 13.3C). When serine is degraded, it is first converted to glycine by serine hydroxymethyltransferase.
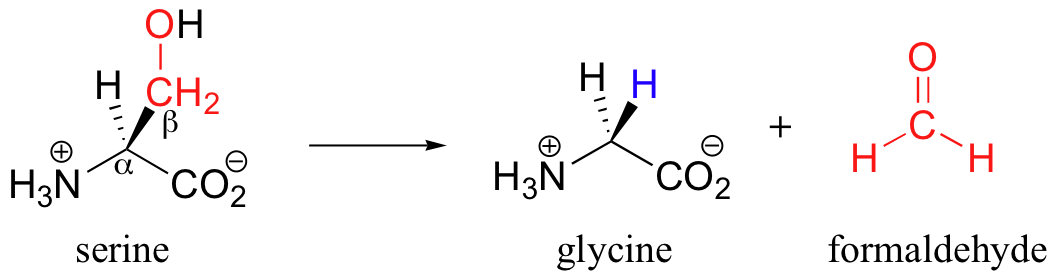
The retro-aldol step occurs from the PLP-serine Schiff base adduct, as the electrons from the broken bond are stabilized by the electron-sink network of the coenzyme. Just like in a standard retroaldol step, these electrons then go on to form a new carbon-hydrogen bond, leading to a PLP-glycine adduct which is freed by imine exchange.
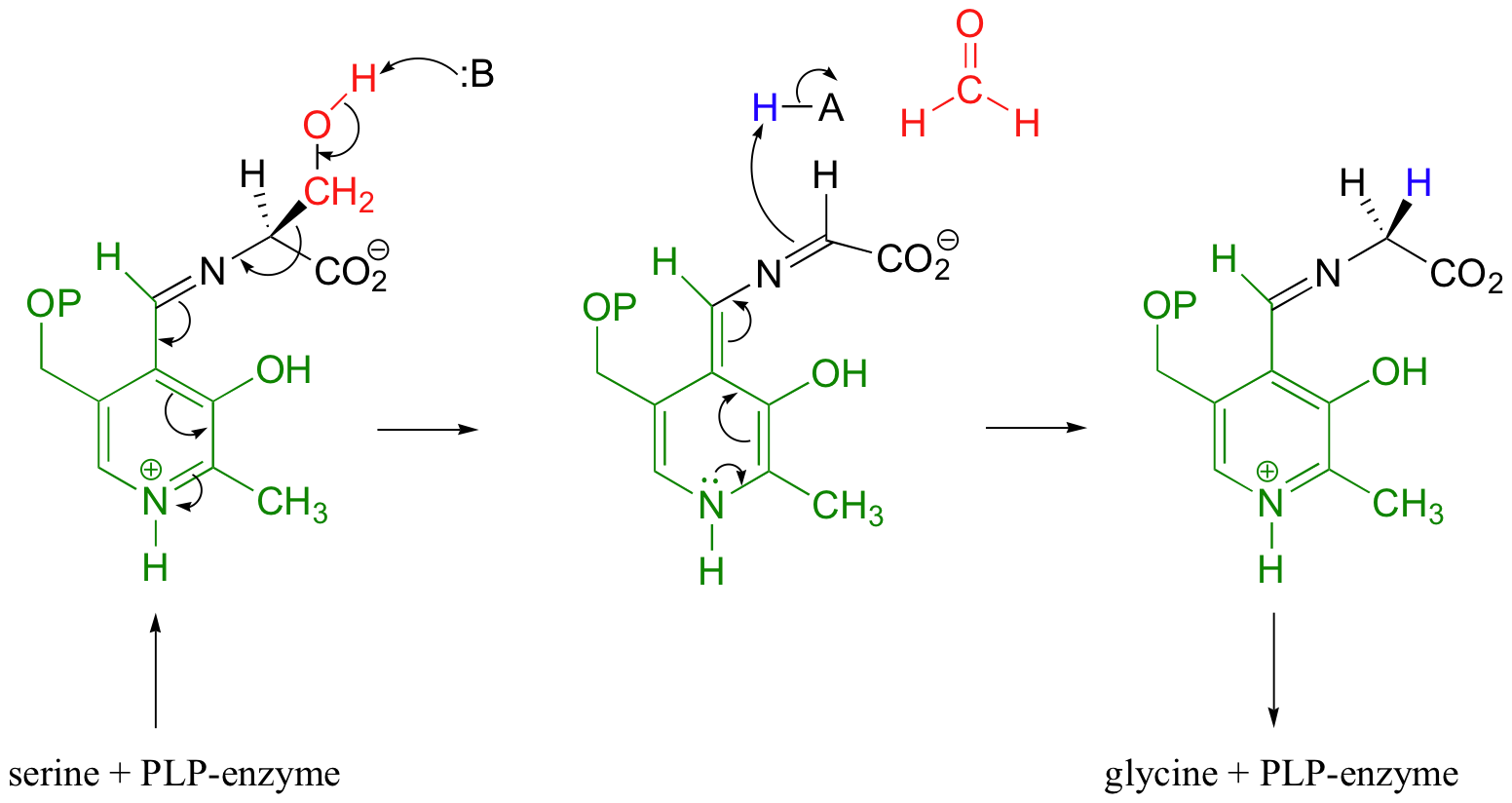
Note that, in this reaction just as in the racemase reaction described previously, the key intermediate is a PLP-stabilized carbanion, or quinonoid.
What happens to the formaldehyde produced in this reaction? Recall from section 11.4D that it is incorporated into the coenzyme tetrahydrofolate to form 5,10-methylenetetrahydrofolate.
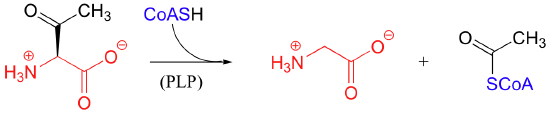
PLP also assists in retro-Claisen cleavage reactions (section 13.4B), such as this step in the degradation of threonine:
14.4E: PLP-dependent transamination reactions
One of the most important steps in the degradation of amino acids is elimination of the amino nitrogen atom in the form of urea, which is excreted in the urine. While the metabolic details of this 'urea cycle' are outside of the scope of this text, what is important to understand is that nitrogen atoms from many of the amino acids must be shuttled first to glutamate before being processed for elimination:
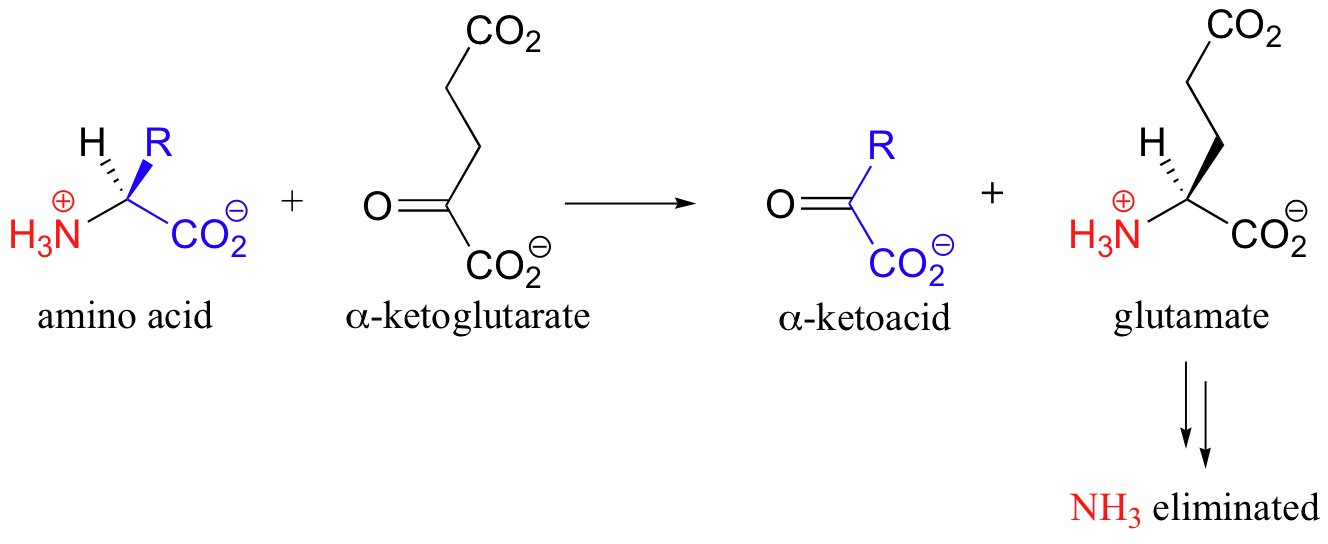
The reaction in which the nitrogen group from an amino acid is transferred to alpha-ketoglutarate is accomplished by PLP-dependent enzymes called transaminases. Once again, the first step is abstraction of the alpha-proton from the PLP-amino acid adduct.
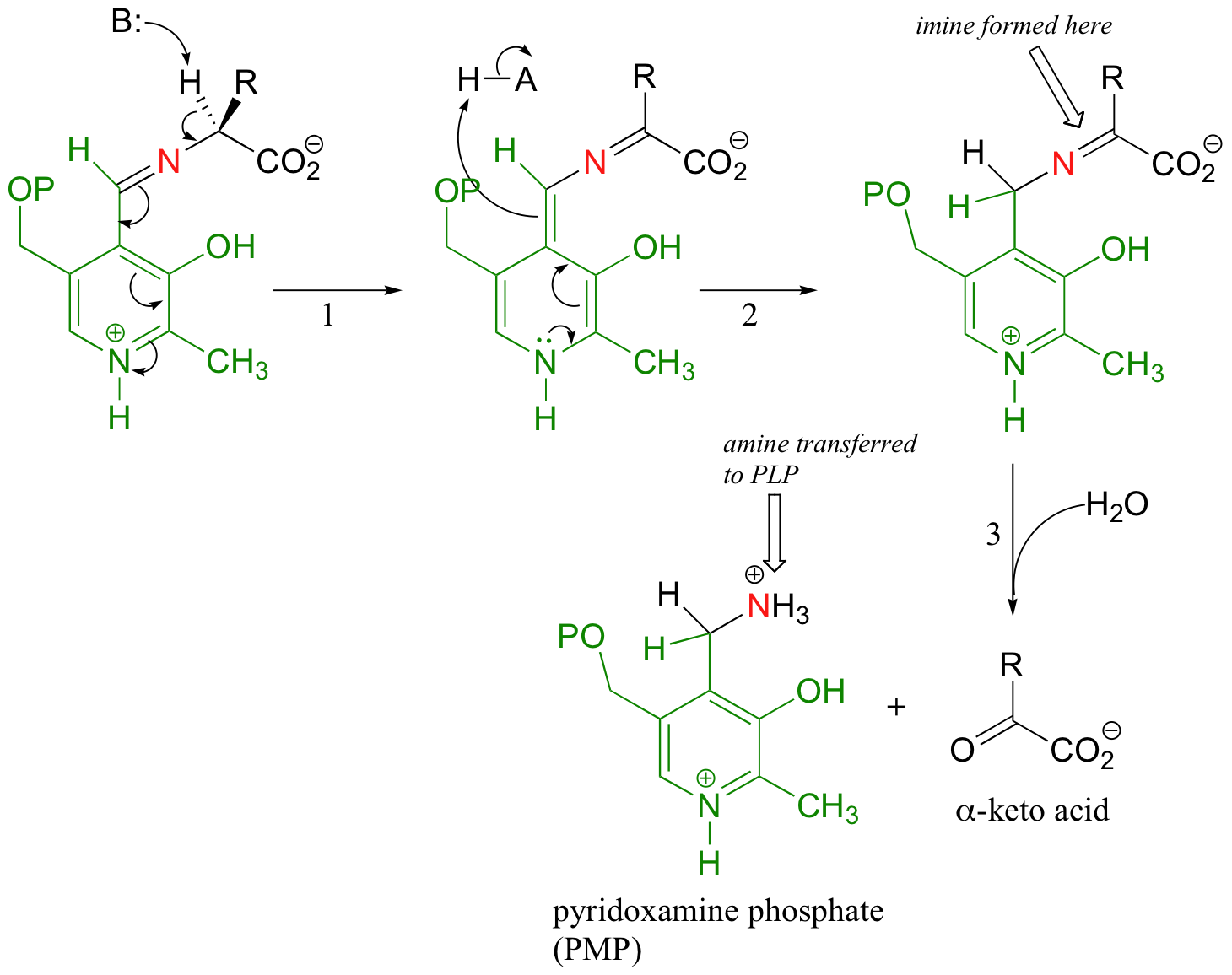
However, in the transaminase reactions this initial deprotonation step is immediately followed by a reprotonation at what was originally the aldehyde carbon of PLP (step 2 above), which results in a new carbon-nitrogen double bond (i.e., an imine) between the a-carbon and the nitrogen of the original amino acid. This imine is then hydrolyzed (step 3 above) - this is the step where the nitrogen is removed from the amino acid to form an alpha-keto acid, which can be degraded further.
The coenzyme, which now carries an amine group and is therefore called pyridoxamine phospate (PMP), next transfers the amine group to alpha-ketoglutarate (to form glutamate) through an exact reversal of the whole process we have just seen.
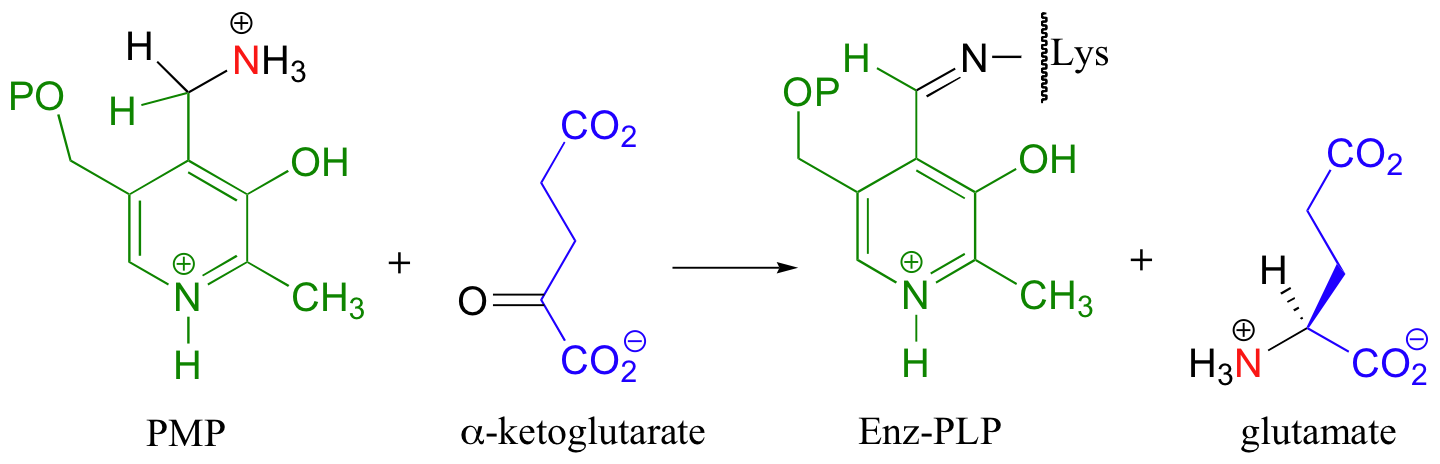
In the overall transaminase reaction, the PLP coenzyme not only provides an electron sink, it also serves as a temporary 'storage space' for a nitrogen atom as it is passed from one amino acid to another.
Exercise 14.5: show a complete, step-by-step mechanism for the second half of the transaminase reaction (transfer of the amine group from PLP to a-ketoglutarate to form glutamate).
Transaminase reactions also function in the biosynthesis direction for alanine, aspartate, and glutamate. Alanine, for example, is synthesized from pyruvate by the transfer of an amino group from glutamate.
14.4F: PLP-dependent beta-elimination and beta-substitution reactions
Two more reaction types in the PLP toolbox are beta-eliminations and beta-substitutions. Serine dehydratase catalyzes a beta-elimination of the hydroxyl group of serine, leading eventually to pyruvate.
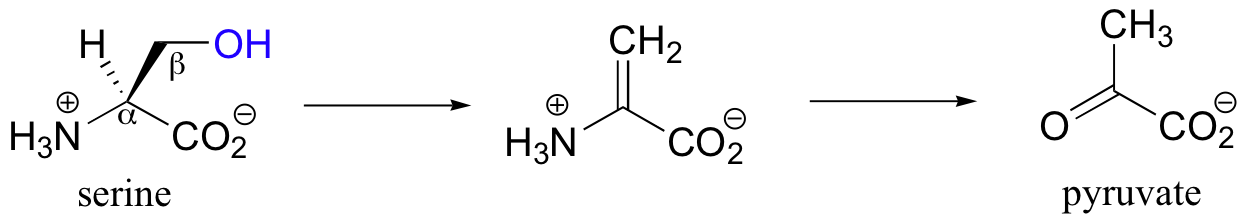
After forming the normal imine linkage with PLP, the a-proton of serine is abstracted by a basic active-site lysine residue (step 1), and the coenzyme stabilizes the conjugate base.
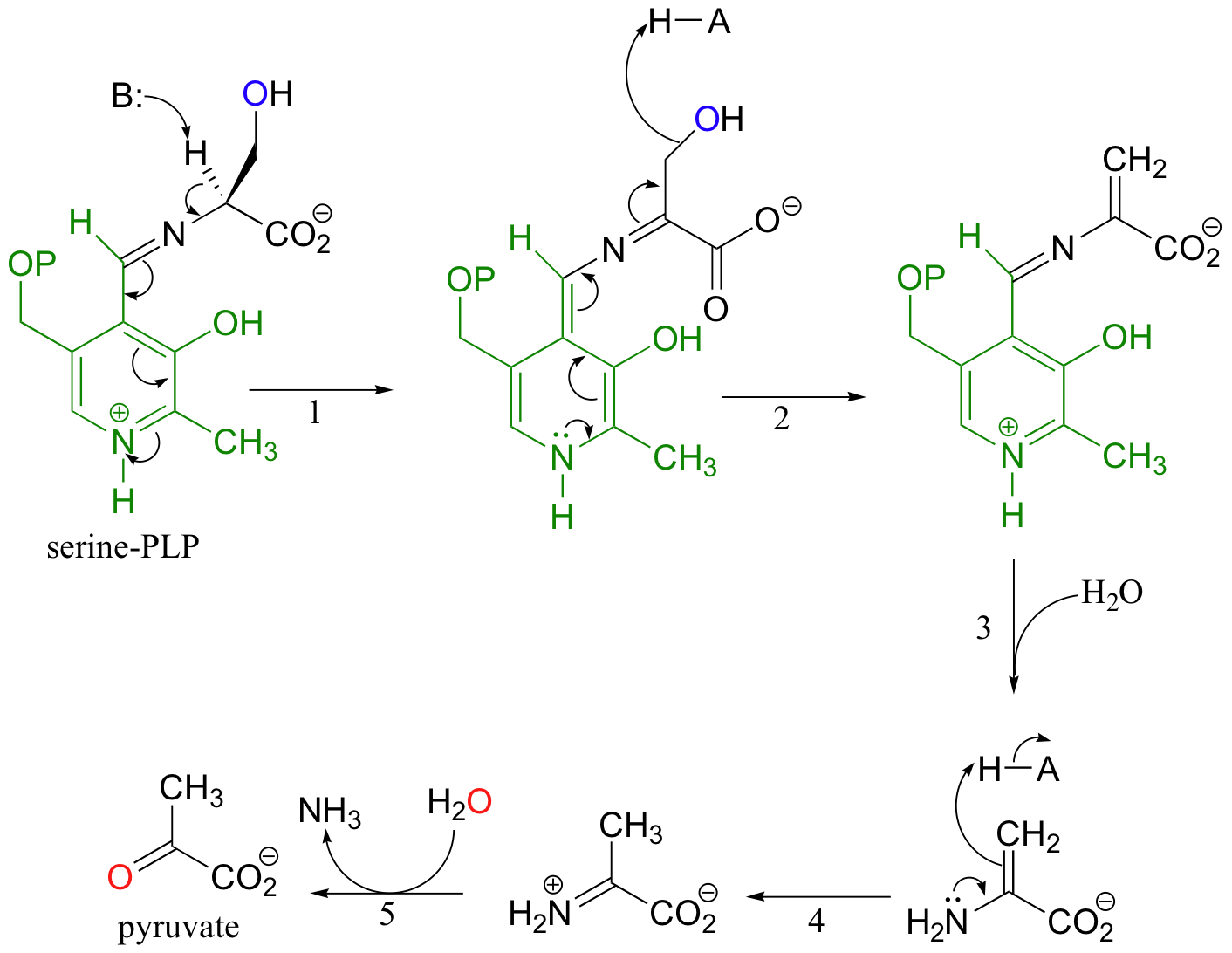
After elimination of water (step 2), the dehydrated substrate is freed from the PLP coenzyme (step 3) by Schiff base transfer, tautomerizes to the imine form (step 4), and then is hydrolyzed to pyruvate (step 5).
A beta-substitution is simply beta-elimination followed directly by the reverse reaction (Michael addition) with a different nucleophile:


As with virtually all PLP-dependent reactions, the coenzyme serves to stabilize the carbanion intermediates.
In many bacteria, the synthesis of cysteine from serine relies upon a PLP-dependent beta-substitution step. In this pathway, serine is first acetylated by acetyl-CoA (an acyl transfer reaction).
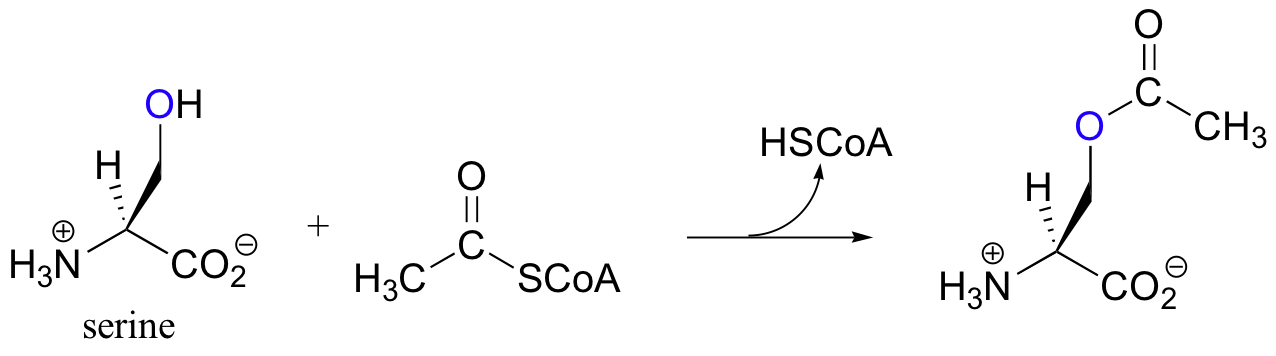
The acetylated serine forms an imine linkage with PLP, then undergoes an elimination (steps 1-2 below) in which the acetyl group is expelled (acetyl is, of course, a much weaker base / better leaving group than a hydroxide - thus the function of the initial serine acetylation step).
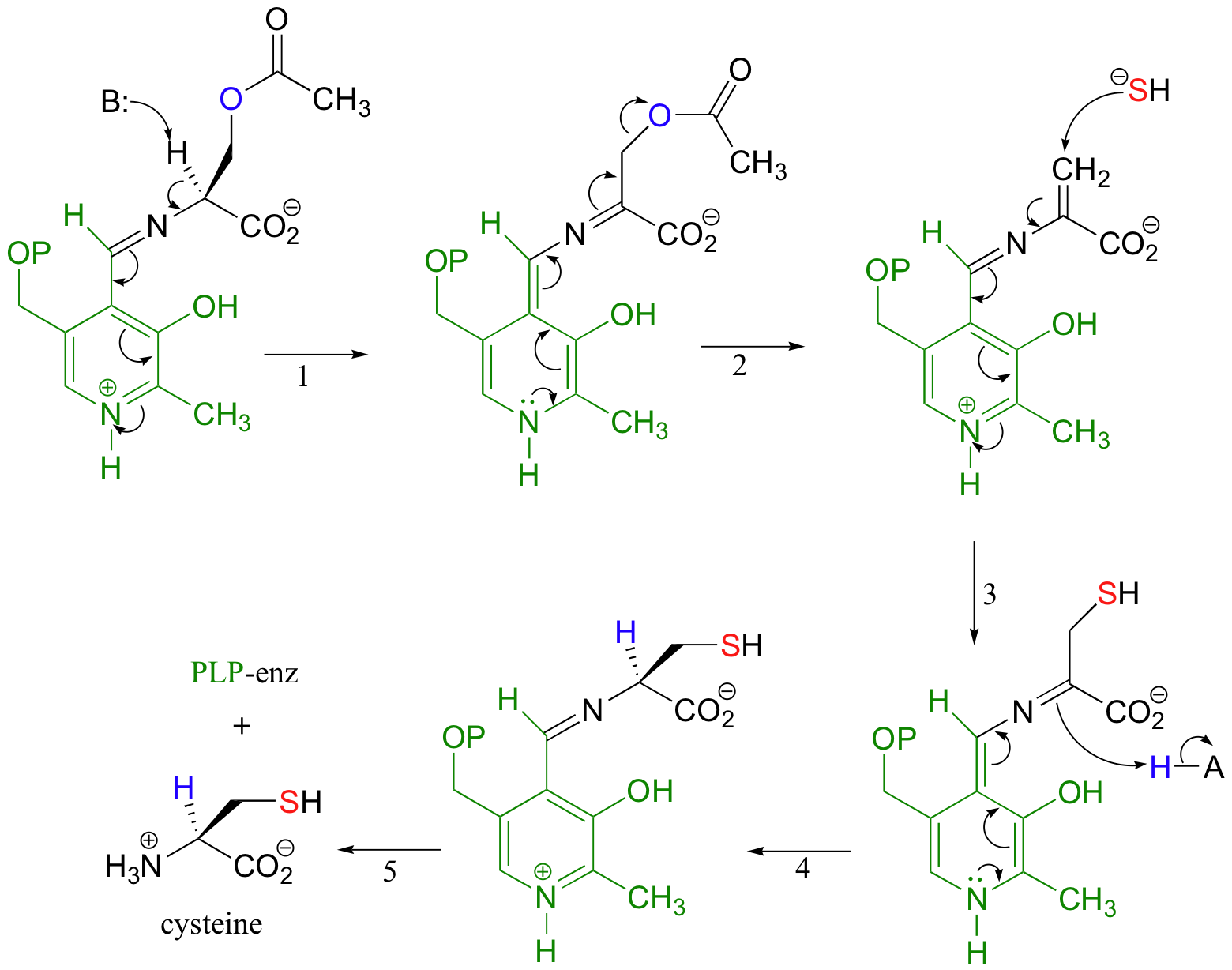
A sulfhydryl ion (SH-) then attacks in a Michael addition (steps 3-4), with the intermediate stabilized again by the electron-sink property of PLP. Finally, the cysteine product is released from PLP (step 5) via an imine exchange reaction with an active site lysine.
14.4G: PLP-dependent gamma-elimination and gamma-substitution reactions
The electron sink capability of PLP allows some enzymes to catalyze eliminations at the gamma (γ) carbon of an amino acid side chain, rather than at the beta-carbon:

The secret to understanding the mechanism of a gamma-elimination is that PLP acts as an electron sink twice - it absorbs the excess electron density from not one but two proton abstractions. As an example, let's look at the cystathionine gamma-lyase reaction, which is part of the methionine degradation pathway.

Cystathionine first links to PLP in the normal way, using the amino group that is furthest away from the sulfur atom. In a familiar step, the alpha-proton is then abstracted by an enzymatic base (step 1), and the electron density is absorbed by PLP.
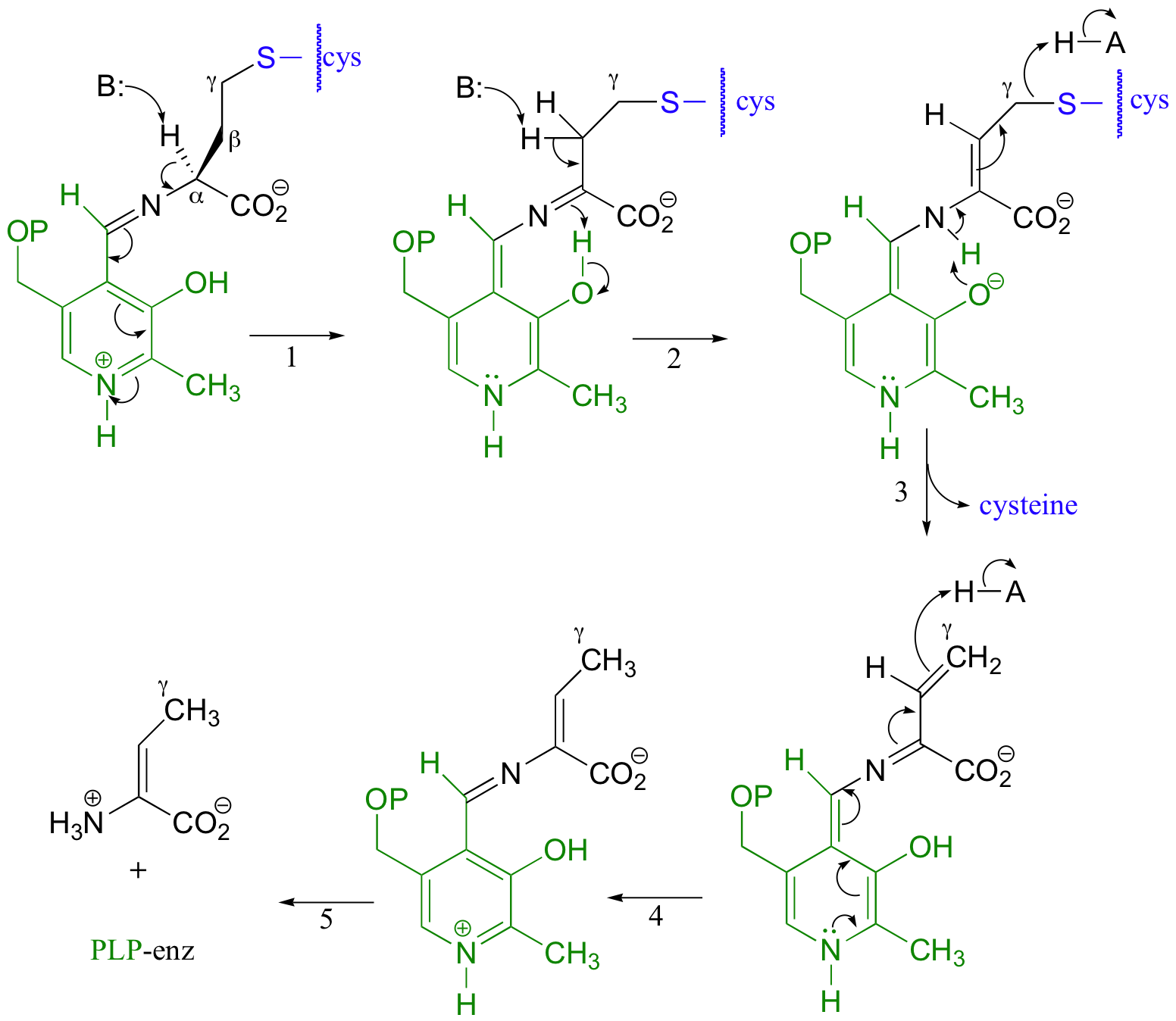
Next comes the new part - before anything happens to the electron density from the first proton abstraction, a second proton, this time from the beta-position on the side chain, is abstracted, forming an enamine intermediate (step 2) This is made possible by the acidic phenolic proton on the pyridoxal ring of PLP. It is this second proton abstraction that is part of a recognizable elimination reaction, as the thiol group (which is actually a cysteine amino acid) leaves and a new pi-bond forms between Cβ and Cγ(step 3). This pi-bond is short-lived, however, as the electron density from the first proton abstraction, which has been 'hiding' in PLP all this time, finally bounces back and protonates Cγ (step 4) With the usual imine transfer to an enzymatic lysine, the final product, 2-aminocrotonate, is released in step 5.
Another related reaction is the PLP-dependant gamma-substitution, which starts off like a gamma-elimination, then reverses itself with the addition of a different nucleophile.

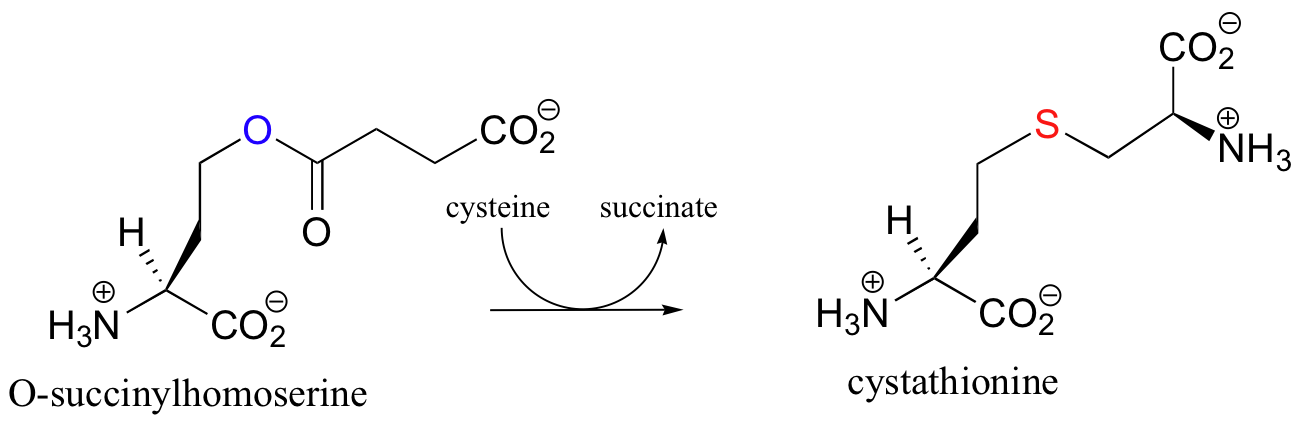
In the synthesis of methionine, cystathionine (the starting compound in the previous gamma-elimination!) is obtained from O-succinyl homoserine and cysteine in a PLP-dependent gamma-substitution.
14.4H: Altering the course of a PLP reaction through site-directed mutagenesis
We have seen how PLP-dependent enzymes catalyze a group of reaction types - racemizations, retroaldols, transamininations, and eliminations - which, despite their apparent diversity, are all characterized by a critical carbanion intermediate that is stabilized by the electron sink of the PLP coenzyme. Given the similarities in the chemistry, it would be reasonable to propose that the active site architecture of these enzymes might also be quite close. This idea was nicely illustrated by an experiment in which site-directed mutagenesis on a single active site amino acid of PLP-dependent alanine racemase was sufficient to turn it into a retro-aldolase when provided with a suitable alternate substrate (J. Am. Chem. Soc. 2003, 125, 10158).

The catalytic base that abstracts the alpha-proton in the alanine racemase reaction is a tyrosine, assisted by a nearby histidine (see figure below). When researchers mutated the tyrosine to an alanine, and substituted beta-hydroxytyrosine for the alanine substrate, a retro-aldol reaction was catalyzed with remarkable efficiency.
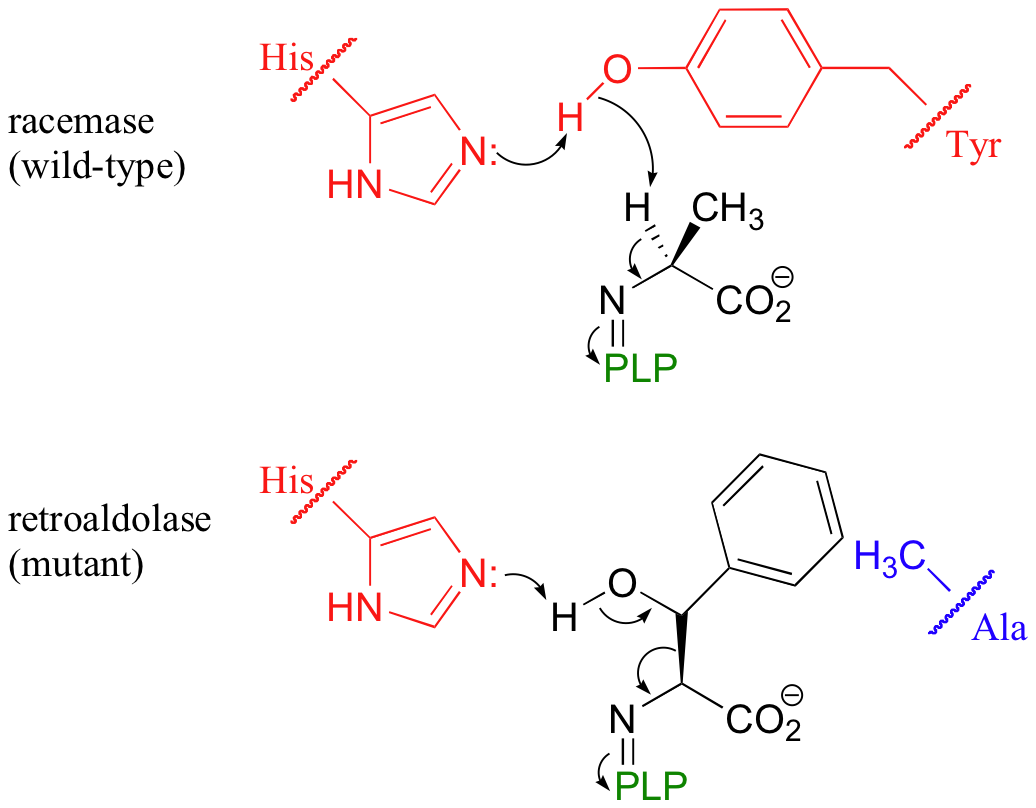
Notice what has happened here: the basic histidine, with no tyrosine to deprotonate because of the mutation, instead abstracts a proton from the beta-hydroxyl group of the new substrate, setting up a retroaldol cleavage. What the researchers did, essentially, was to swap a beta-hydroxy amino acid substrate (capable of undergoing retroaldol cleavage) for the normal alanine substrate, then reposition the active site basic group so that a different acidic proton was abstracted. That was all it took to change a racemase into a retroaldolase, because the necessary electron sink system was all left in place. Why did they use the unnatural amino acid beta-hydroxy tyrosine for the retro-aldol substrate rather than threonine, which also has a beta-hydroxyl group and is closer in structure to alanine? The researchers figured that the phenyl ring of beta-hydroxy tyrosine would fit nicely in the space left empty due to the removal of the enzymatic tyrosine from the active site. To reiterate: the point of this experiment was to demonstrate how similar the chemistry being catalyzed by different PLP-dependent enzymes really is - and, as a corollary, how similar the active sites are. Even though one would not normally consider a racemization to be closely related to a retroaldol cleavage, the mechanistic themes are very close, as evidenced by researchers' ability to change the reaction product by making a single active site mutation.