
27.2: Instrumentos para Cromatografía de Gases
- Page ID
- 79009

En cromatografía de gases (GC) inyectamos la muestra, que puede ser un gas o un líquido, en una fase móvil gaseosa (a menudo llamada gas portador). La fase móvil transporta la muestra a través de una columna empaquetada o capilar que separa los componentes de la muestra en función de su capacidad de partición entre la fase móvil y la fase estacionaria. La Figura 27.2.1 muestra un ejemplo de un cromatógrafo de gases típico, el cual consta de varios componentes clave: un suministro de gas comprimido para la fase móvil; un inyector calentado, que volatiliza rápidamente los componentes en una muestra líquida; una columna, que se coloca dentro de un horno cuya temperatura nosotros puede controlar durante la separación; y un detector para monitorear el eluyente a medida que sale de la columna. Consideremos cada uno de estos componentes.

Fase Móvil
Las fases móviles más comunes para la cromatografía de gases son He, Ar y N 2, las cuales tienen la ventaja de ser químicamente inertes tanto hacia la muestra como hacia la fase estacionaria. La elección del gas portador a menudo está determinada por las necesidades del detector del instrumento. Para una columna empaquetada, la velocidad de la fase móvil suele ser de 25—150 ml/min. El caudal típico para una columna capilar es de 1—25 ml/min.
Introducción a la muestra
Tres factores determinan cómo se introduce una muestra en el cromatógrafo de gases. Primero, todos los componentes de la muestra deben ser volátiles. Segundo, los analitos deben estar presentes en una concentración apropiada. Finalmente, el proceso físico de inyección de la muestra no debe degradar la separación. Cada una de estas necesidades se considera en esta sección.
Preparación de una muestra volátil
No todas las muestras pueden inyectarse directamente en un cromatógrafo de gases. Para desplazarse por la columna, los componentes de la muestra deben ser suficientemente volátiles. Un soluto de baja volatilidad, por ejemplo, puede ser retenido por la columna y continuar eluyendo durante el análisis de muestras posteriores. Un soluto no volátil se condensará en la parte superior de la columna, degradando el rendimiento de la columna.
Un enfoque atractivo para aislar analitos es una microextracción en fase sólida (SPME). En un enfoque, que se ilustra en la Figura 27.2.2 , se coloca una fibra de sílice fundida dentro de una aguja de jeringa. La fibra, que se recubre con una película delgada de un material adsorbente, como el polidimetilsiloxano, se baja en la muestra presionando un émbolo y se expone a la muestra por un tiempo predeterminado. Después de retirar la fibra en la aguja, se transfiere al cromatógrafo de gases para su análisis.

Dos métodos adicionales para aislar analitos volátiles son un muestreo de purga y trampa y espacio de cabeza. En una purga y trampa, burbujeamos un gas inerte, como He o N 2, a través de la muestra, liberando o purgando los compuestos volátiles. Estos compuestos son transportados por el gas de purga a través de una trampa que contiene un material absorbente, como Tenax, donde son retenidos. El calentamiento de la trampa y el lavado a contracorriente con gas portador transfiere los compuestos volátiles al cromatógrafo de gases. En el muestreo del espacio de cabeza colocamos la muestra en un vial cerrado con un espacio aéreo superpuesto. Después de dejar tiempo para que los analitos volátiles se equilibren entre la muestra y el aire suprayacente, utilizamos una jeringa para extraer una porción de la fase de vapor e inyectarla en el cromatógrafo de gases. Alternativamente, podemos muestrear el espacio de cabeza con un SPME.
La desorción térmica es un método útil para liberar analitos volátiles de sólidos. Colocamos una porción del sólido en un tubo de acero inoxidable revestido de vidrio. Después de purgar con gas portador para eliminar cualquier O 2 que pudiera estar presente, calentamos la muestra. Los analitos volátiles son barridos del tubo por un gas inerte y transportados al GC. Debido a que la volatilización no es un proceso rápido, los analitos volátiles a menudo se concentran en la parte superior de la columna enfriando la entrada de la columna por debajo de la temperatura ambiente, un proceso conocido como enfoque criogénico. Una vez completada la volatilización, la entrada de la columna se calienta rápidamente, liberando los analitos para que viajen a través de la columna.
La razón para eliminar O 2 es evitar que la muestra sufra una reacción de oxidación cuando se calienta.
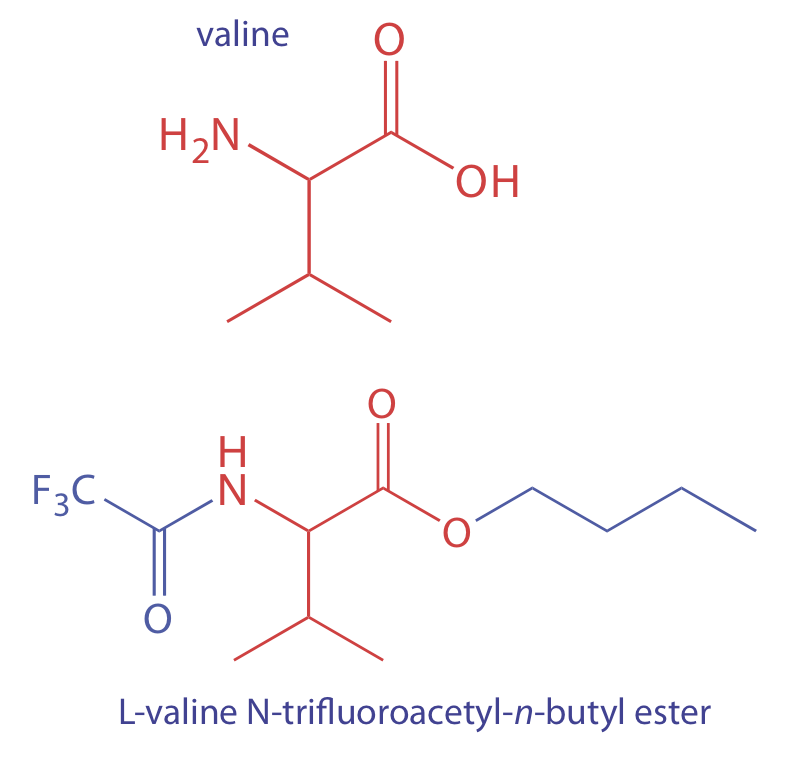
Para analizar un analito no volátil debemos convertirlo en una forma volátil. Por ejemplo, los aminoácidos no son lo suficientemente volátiles para analizarlos directamente por cromatografía de gases. La reacción de un aminoácido, como la valina, con 1-butanol y cloruro de acetilo produce un aminoácido esterificado. El tratamiento posterior con ácido trifluoroacético da el derivado volátil del éster N-trifluoroacetil- n-butílico del aminoácido.
Ajuste de la concentración del analito
En un analito la concentración es demasiado pequeña para dar una señal adecuada, entonces debemos concentrar el analito antes de inyectar la muestra en el cromatógrafo de gases. Un beneficio secundario de muchos métodos de extracción es que a menudo concentran los analitos. Los materiales orgánicos volátiles aislados de una muestra acuosa mediante una purga y trampa, por ejemplo, se concentran tanto como\(1000 \times\).
Si un analito está demasiado concentrado, es fácil sobrecargar la columna, lo que resulta en un frente de pico y una separación deficiente. Además, la concentración del analito puede exceder la respuesta lineal del detector. Inyectar menos muestra o diluir la muestra con un disolvente volátil, como el cloruro de metileno, son dos posibles soluciones a este problema.
Inyectar la muestra
En el Capítulo 26 examinamos varias explicaciones de por qué la banda de un soluto aumenta de ancho a medida que pasa por la columna, un proceso que llamamos ensanchamiento de banda. También introducimos una fuente adicional de ensanchamiento de banda si no conseguimos inyectar la muestra en el volumen mínimo posible de fase móvil. Hay dos fuentes principales de este ensanchamiento de banda precolumna: inyectar la muestra en una corriente móvil de fase móvil e inyectar una muestra líquida en lugar de una muestra gaseosa. El diseño del inyector de un cromatógrafo de gases ayuda a minimizar estos problemas.
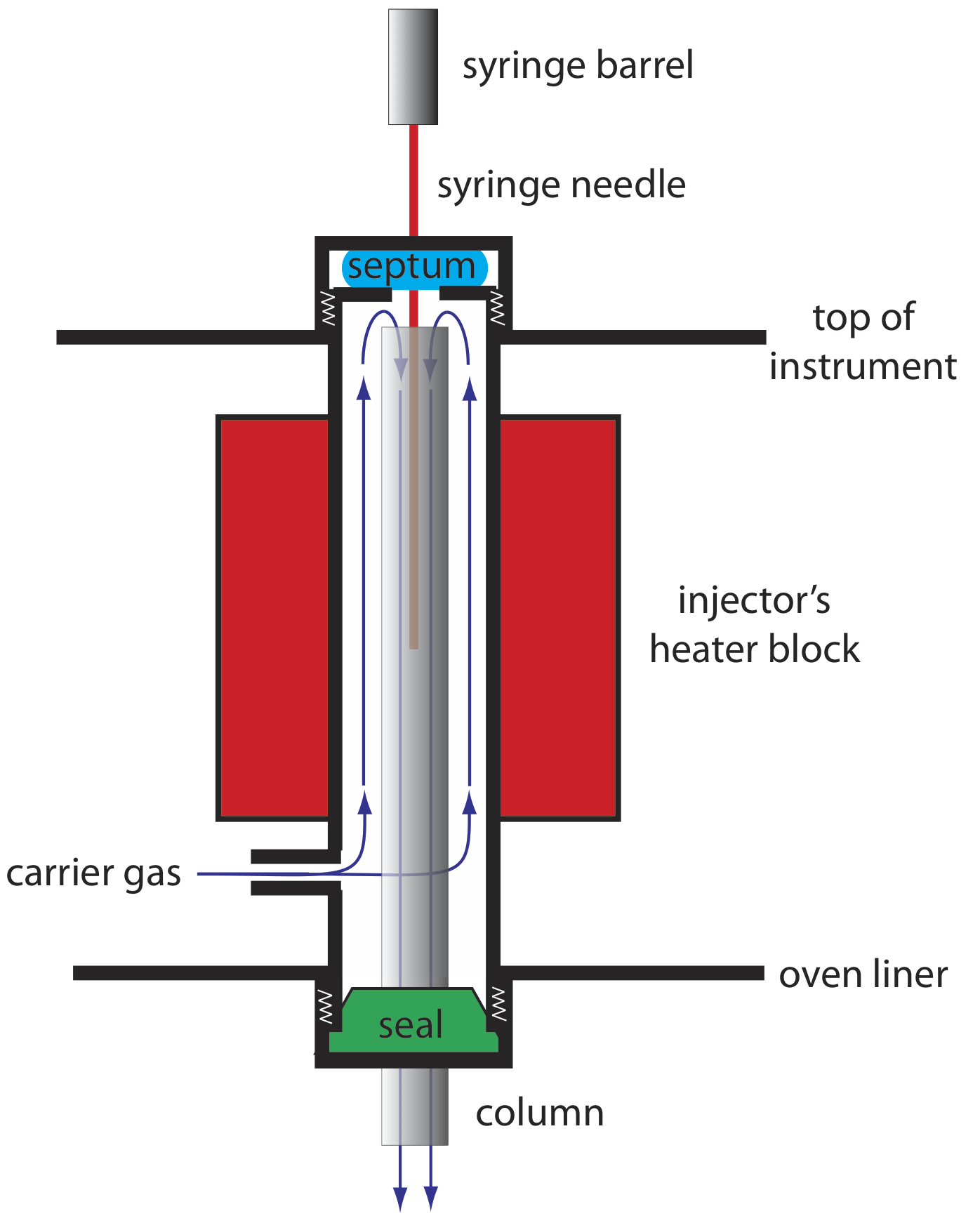
Un ejemplo de un puerto de inyección simple para una columna empaquetada se muestra en la Figura 27.2.3 . La parte superior de la columna encaja dentro de un bloque inyector calentado, con gas portador entrando desde la parte inferior. La muestra se inyecta a través de un tabique de goma usando una jeringa de microlitros, como la que se muestra en la Figura 27.2.4 . Inyectar la muestra directamente en la columna minimiza el ensanchamiento de banda porque mezcla la muestra con la menor cantidad posible de gas portador. El bloque inyector se calienta a una temperatura de al menos 50 o C por encima del punto de ebullición del soluto menos volátil, lo que asegura una rápida vaporización de los componentes de la muestra.

Debido a que el volumen de una columna capilar es significativamente menor que el de una columna empaquetada, requiere un estilo diferente de inyector para evitar sobrecargar la columna con muestra. La figura 27.2.5 muestra un diagrama esquemático de un inyector típico de split/splitless para su uso con una columna capilar.
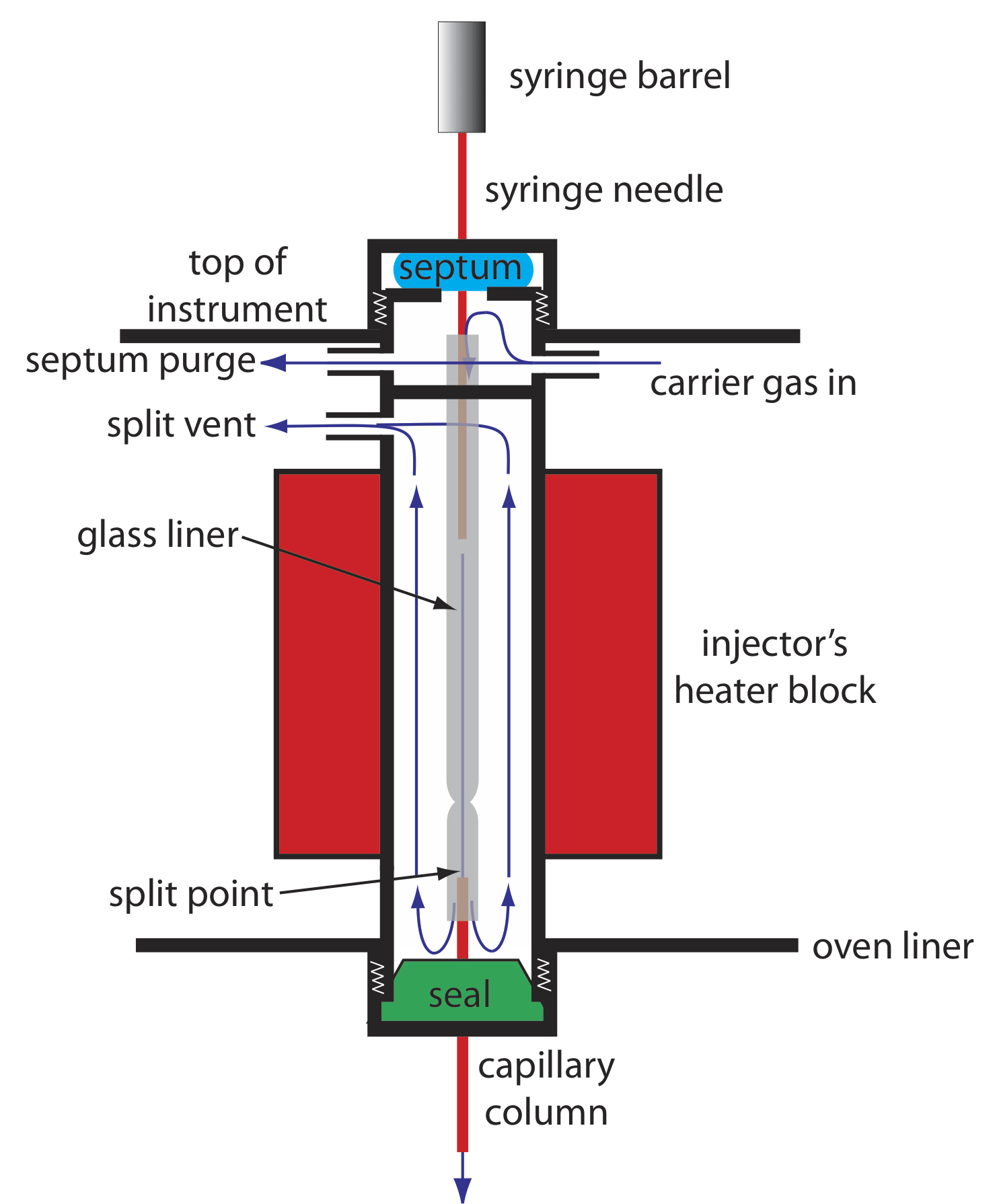
En una inyección dividida inyectamos la muestra a través de un septo de goma usando una jeringa de microlitro. En lugar de inyectar la muestra directamente en la columna, se inyecta en un revestimiento de vidrio donde se mezcla con el gas portador. En el punto de división, una pequeña fracción del gas portador y la muestra ingresa a la columna capilar con el resto saliendo por el respiradero dividido. Al controlar el caudal del gas portador a medida que ingresa al inyector, y su caudal a través de la purga del tabique y la ventilación dividida, podemos controlar la fracción de muestra que ingresa a la columna capilar, típicamente 0.1— 10%.
Por ejemplo, si el caudal de gas portador es de 50 ml/min, y los caudales para la purga del tabique y el respiradero dividido son de 2 ml/min y 47 ml/min, respectivamente, entonces el caudal a través de la columna es de 1 ml/min (= 50 — 2 — 47). La proporción de muestra que ingresa a la columna es 1/50, o 2%.
En una inyección sin división, que es útil para el análisis de trazas, cerramos la ventilación dividida y permitimos que todo el gas portador que pasa a través del revestimiento de vidrio ingrese a la columna, esto permite que prácticamente toda la muestra ingrese a la columna. Debido a que el caudal a través del inyector es bajo, el ensanchamiento significativo de la banda precolumna es un problema. El mantenimiento de la temperatura de la columna aproximadamente 20—25 o C por debajo del punto de ebullición del disolvente permite que el disolvente se condense a la entrada de la columna capilar, formando una barrera que atrapa los solutos. Después de permitir que los solutos se concentren, se incrementa la temperatura de la columna y se inicia la separación.
Para muestras que se descomponen fácilmente, puede ser necesaria una inyección en columna. En este método la muestra se inyecta directamente en la columna sin calentar. Luego se incrementa la temperatura de la columna, volatilizando la muestra con una temperatura tan baja como sea práctico.
Control de temperatura
El control de la temperatura de la columna es crítico para lograr una buena separación cuando se usa cromatografía de gases. Por esta razón la columna se coloca dentro de un horno termostático (ver Figura 27.2.1 ). En una separación isotérmica mantenemos la columna a una temperatura constante. Para aumentar la interacción entre los solutos y la fase estacionaria, la temperatura generalmente se establece ligeramente por debajo de la del soluto de menor punto de ebullición.
Una dificultad con una separación isotérmica es que una temperatura que favorece la separación de un soluto de bajo punto de ebullición puede conducir a un tiempo de retención inaceptablemente largo para un soluto de mayor punto de ebullición. La programación de temperatura proporciona una solución a este problema. Al inicio del análisis establecemos la temperatura inicial de la columna por debajo de la del soluto de menor punto de ebullición. A medida que avanza la separación, aumentamos lentamente la temperatura a una velocidad uniforme o en una serie de pasos.
Detectores para cromatografía de gases
La parte final de un cromatógrafo de gases es el detector. El detector ideal tiene varias características deseables: un bajo límite de detección, una respuesta lineal en un amplio rango de concentraciones de solutos (lo que facilita el trabajo cuantitativo), sensibilidad para todos los solutos o selectividad para una clase específica de solutos, y una insensibilidad a un cambio en el caudal o temperatura.
Detector de Conductividad Térmica (TCD)
Uno de los primeros detectores de cromatografía de gases aprovecha la conductividad térmica de la fase móvil. A medida que la fase móvil sale de la columna pasa sobre un filamento de alambre de tungsteno-renio (ver Figura 27.2.6 ). La resistencia eléctrica del filamento depende de su temperatura, la cual, a su vez, depende de la conductividad térmica de la fase móvil. Debido a su alta conductividad térmica, el helio es la fase móvil de elección cuando se utiliza un detector de conductividad térmica (TCD).
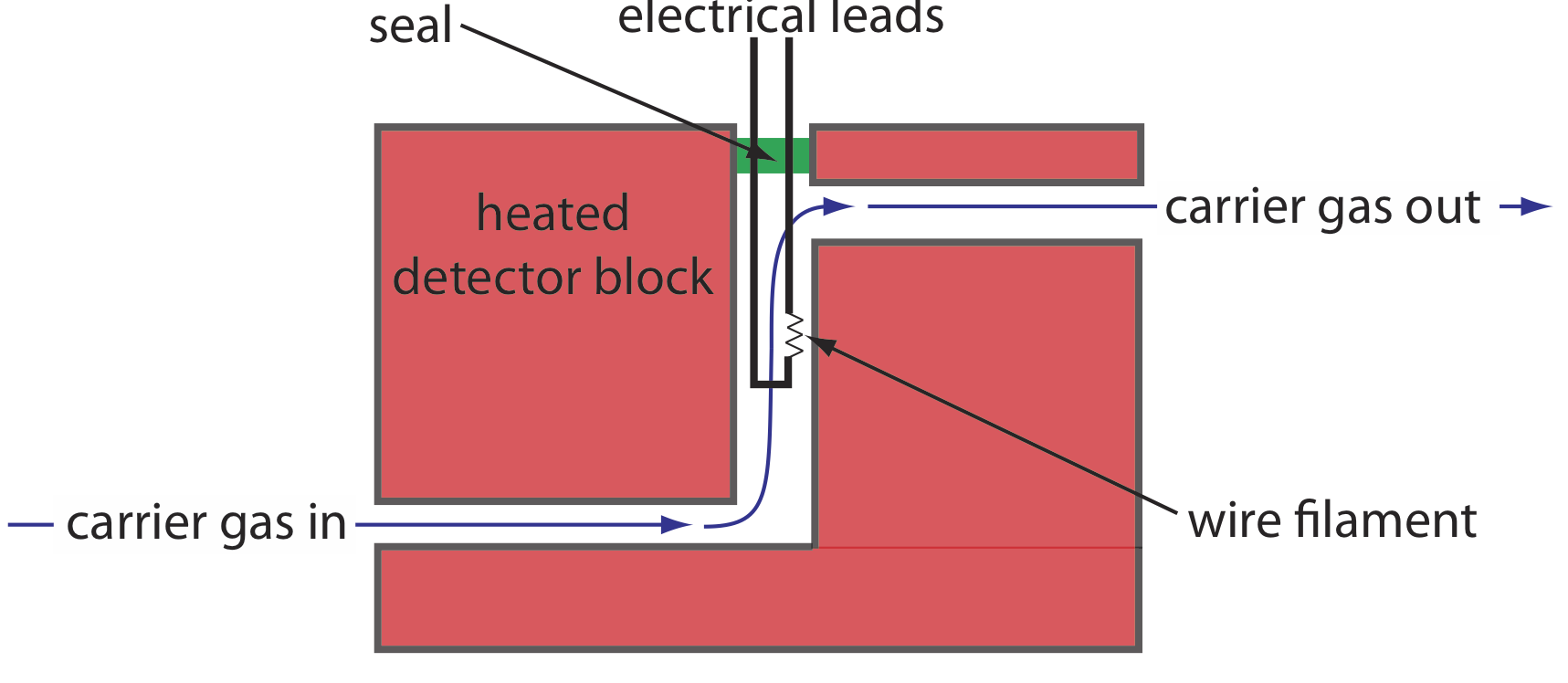
La conductividad térmica, como su nombre indica, es una medida de la facilidad con la que una sustancia conduce el calor. Un gas con una alta conductividad térmica aleja el calor del filamento y, por lo tanto, lo enfría más rápidamente que un gas con baja conductividad térmica.
Cuando un soluto eluye de la columna, la conductividad térmica de la fase móvil en la celda TCD disminuye y la temperatura del filamento de alambre, y por lo tanto su resistencia, aumenta. Una celda de referencia, a través de la cual solo pasa la fase móvil, corrige cualquier variación dependiente del tiempo en el caudal, la presión o la energía eléctrica, todas las cuales afectan la resistencia del filamento.
Debido a que todos los solutos afectan la conductividad térmica de la fase móvil, el detector de conductividad térmica es un detector universal. Otra ventaja es la respuesta lineal del TCD sobre un rango de concentración que abarca 10 4 —10 5 órdenes de magnitud. El detector también es no destructivo, lo que nos permite recuperar analitos usando una trampa fría posdetector. Una desventaja significativa del detector TCD es su escaso límite de detección para la mayoría de los analitos.
Detector de Ionización de Llama (FID)
La combustión de un compuesto orgánico en una llama H 2 /aire da como resultado una llama que contiene electrones y cationes orgánicos, presumiblemente CHO +. La aplicación de un potencial de aproximadamente 300 voltios a través de la llama crea una pequeña corriente de aproximadamente 10 —9 a 10 —12 amperios. Cuando se amplifica, esta corriente proporciona una señal analítica útil. Esta es la base del popular detector de ionización de llama, un diagrama esquemático del cual se muestra en la Figura 27.2.7 .
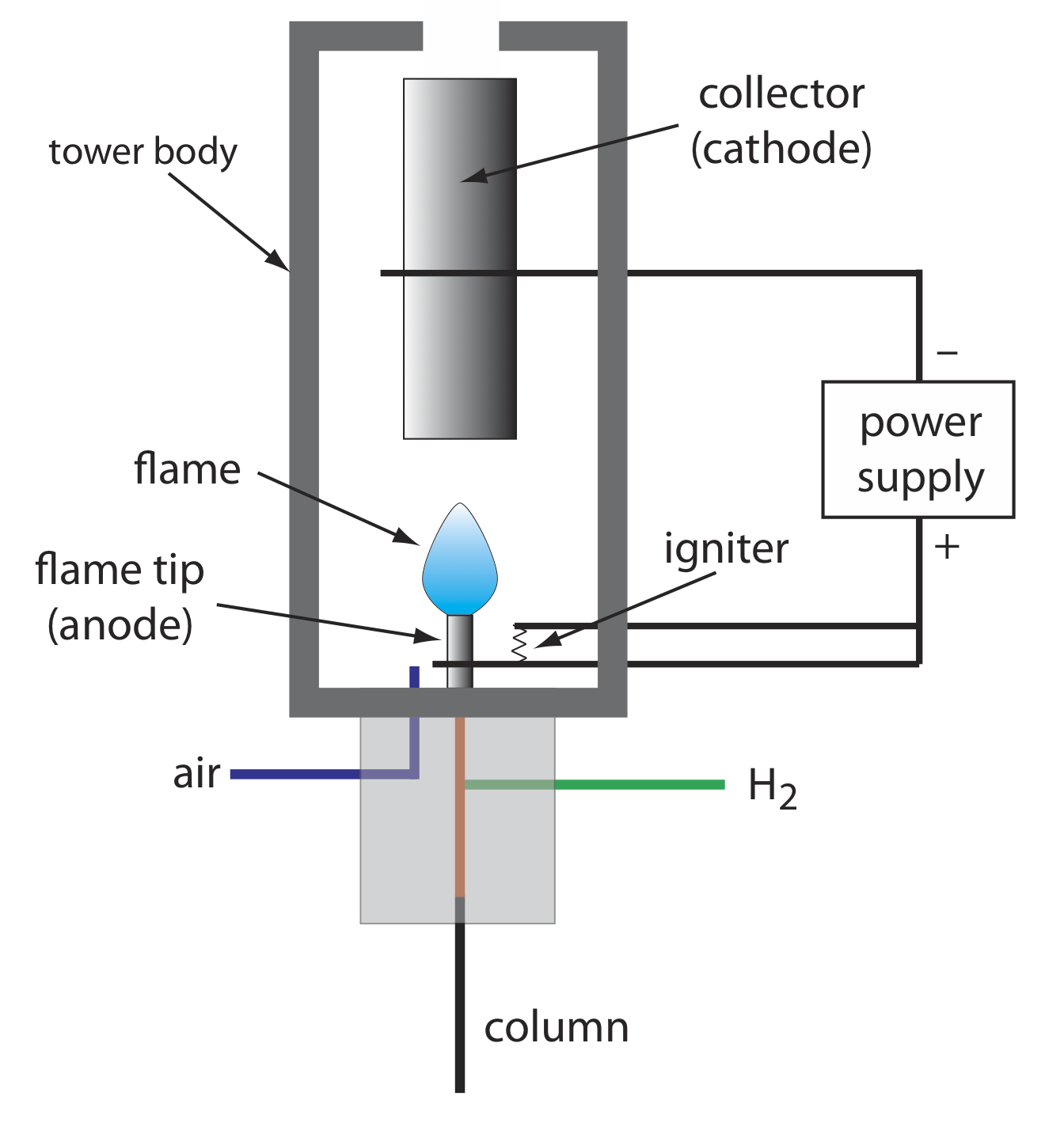
La mayoría de los átomos de carbono, excepto los de los grupos carbonilo y carboxílico, generan una señal, lo que convierte al FID en un detector casi universal para compuestos orgánicos. La mayoría de los compuestos inorgánicos y muchos gases, como H 2 O y CO 2, no se detectan, lo que convierte al detector FID en un detector útil para el análisis de analitos orgánicos en muestras ambientales atmosféricas y acuosas. Las ventajas del FID incluyen un límite de detección que es aproximadamente de dos a tres órdenes de magnitud menor que el de un detector de conductividad térmica, y una respuesta lineal superior a 10 6 —10 7 órdenes de magnitud en la cantidad de analito inyectado. La muestra, por supuesto, se destruye cuando se utiliza un detector de ionización de llama.
Detector de captura de electrones (ECD)
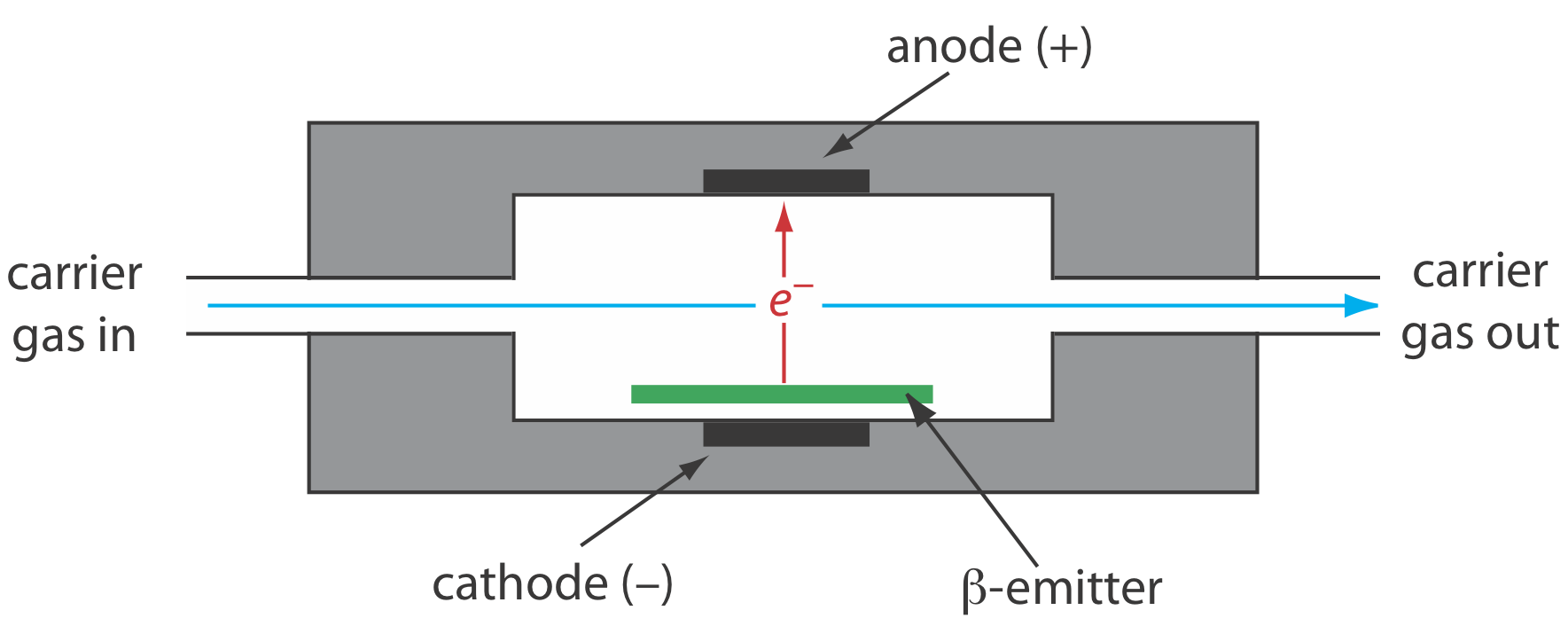
El detector de captura de electrones es un ejemplo de un detector selectivo. Como se muestra en la Figura 27.2.8 , el detector consiste en un\(\beta\) -emisor, tal como 63 Ni. Los electrones emitidos ionizan la fase móvil, generalmente N 2, generando una corriente estacionaria entre un par de electrodos. Cuando un soluto con una alta afinidad para capturar electrones eluye de la columna, la corriente disminuye, lo que sirve como señal. El ECD es altamente selectivo hacia solutos con grupos funcionales electronegativos, como halógenos y grupos nitro, y es relativamente insensible a aminas, alcoholes e hidrocarburos. Aunque su límite de detección es excelente, su rango lineal se extiende solo en aproximadamente dos órdenes de magnitud.
Una\(\beta\) partícula es un electrón.
Espectrómetro de masas (MS)
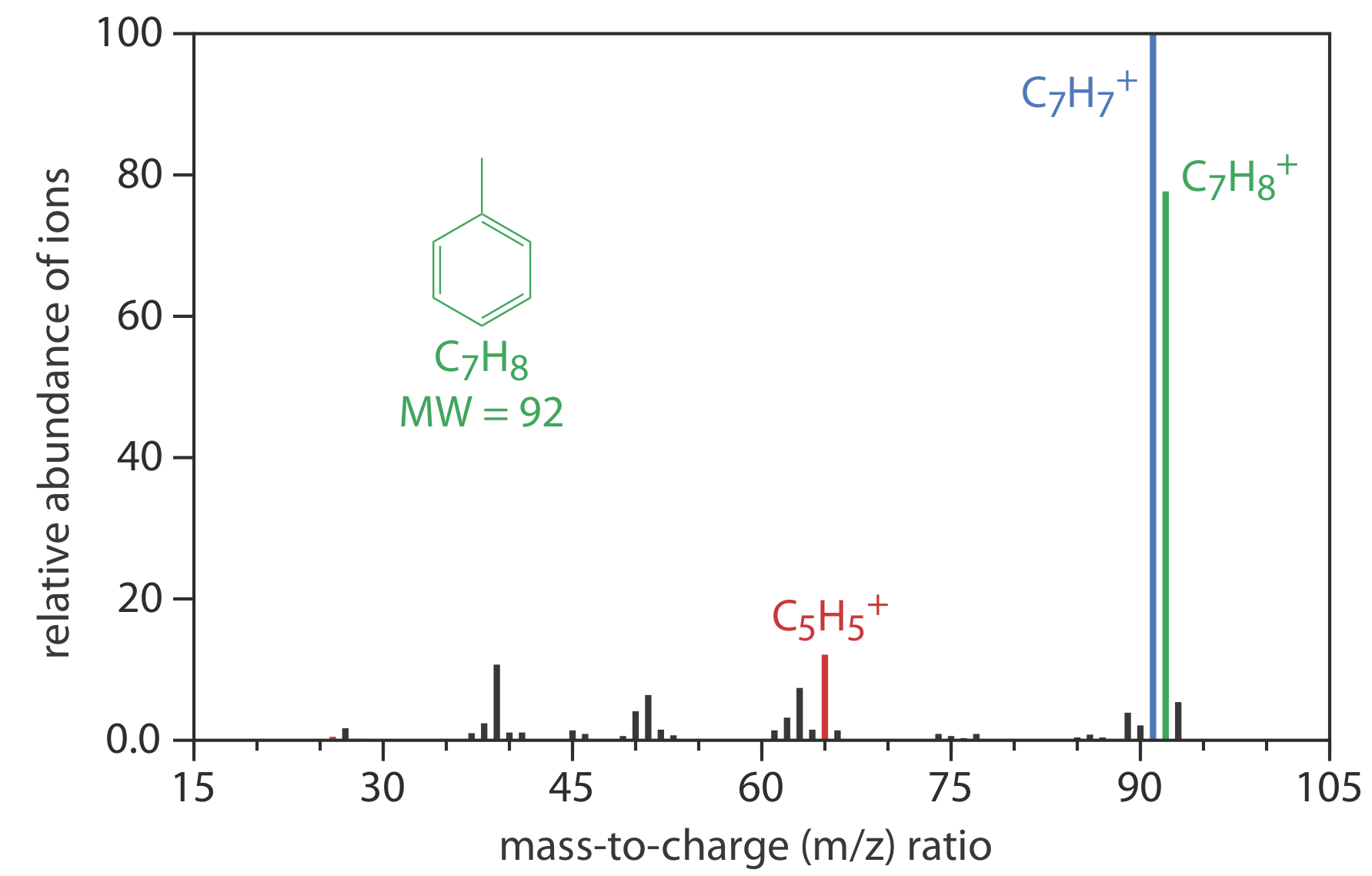
Un espectrómetro de masas es un instrumento que ioniza una molécula gaseosa utilizando suficiente energía para que el ion resultante se descomponga en iones más pequeños. Debido a que estos iones tienen diferentes relaciones de masa a carga, es posible separarlos usando un campo magnético o un campo eléctrico. El espectro de masas resultante contiene información tanto cuantitativa como cualitativa sobre el analito. La Figura 27.2.9 muestra un espectro de masas para tolueno.
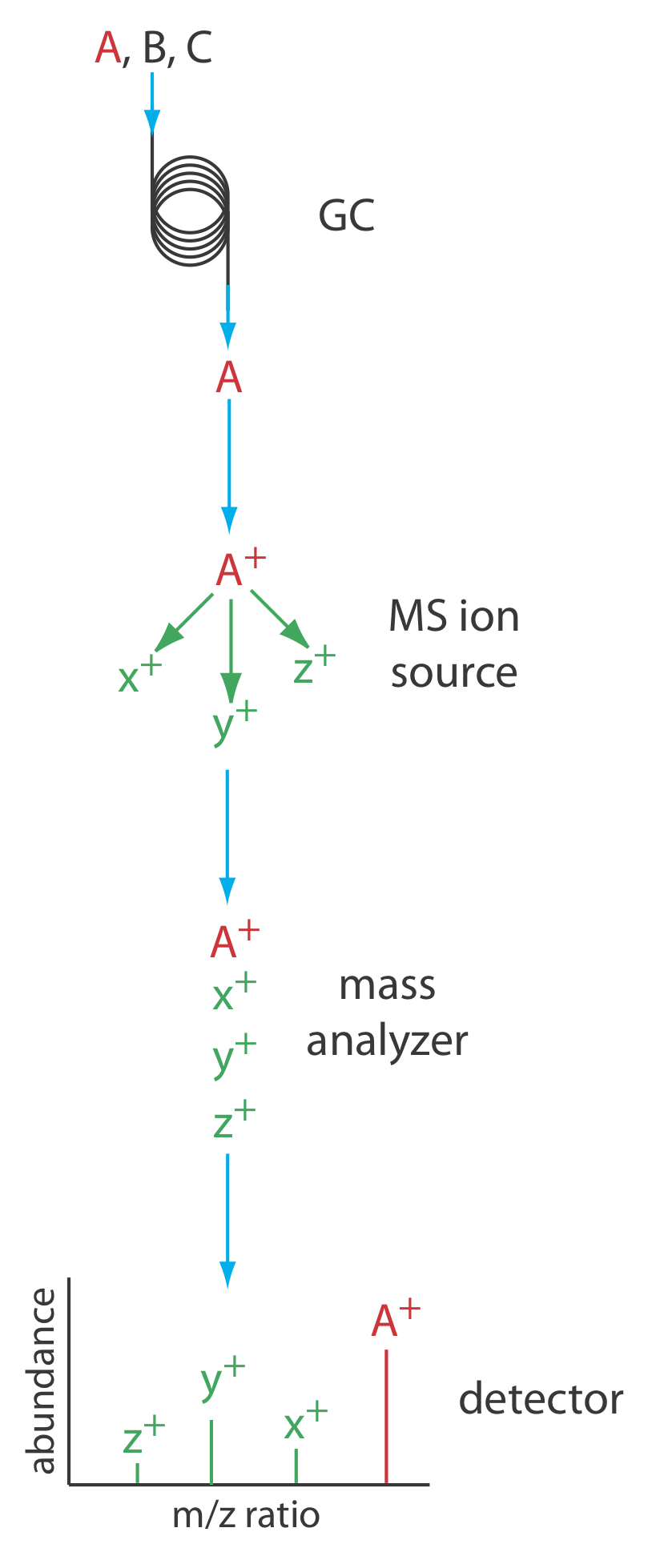
La Figura 27.2.10 muestra un diagrama de bloques de un instrumento típico de cromatografía de gases-espectrómetro de masas (GC-MS). El efluente de la columna ingresa a la fuente de iones del espectrómetro de masas de manera que elimina la mayoría del gas portador. En la cámara de ionización, las moléculas restantes, una mezcla de gas portador, disolvente y solutos, experimentan ionización y fragmentación. El analizador de masas del espectrómetro de masas separa los iones por su relación masa-carga y un detector cuenta los iones y muestra el espectro de masas.
Existen varias opciones para monitorear un cromatograma cuando se usa un espectrómetro de masas como detector. El método más común es escanear continuamente todo el espectro de masas y reportar la señal total para todos los iones que llegan al detector durante cada exploración. Este escaneo de iones totales proporciona detección universal para todos los analitos. Podemos lograr cierto grado de selectividad monitoreando una o más relaciones específicas de masa a carga, un proceso llamado monitoreo de iones selectivos. Un espectrómetro de masas proporciona excelentes límites de detección, típicamente de 25 fg a 100 pg, con un rango lineal de 10 5 órdenes de magnitud. Debido a que registramos continuamente el espectro de masas del eluyente de la columna, podemos retroceder y examinar el espectro de masas para cualquier incremento de tiempo. Esta es una clara ventaja para GC-MS porque podemos usar el espectro de masas para ayudar a identificar los componentes de una mezcla.
Otros Detectores
Dos detectores adicionales son similares en diseño a un detector de ionización de llama. En el detector fotométrico de llama, la emisión óptica de fósforo y azufre proporciona un detector selectivo para compuestos que contienen estos elementos. El detector termiónico responde a compuestos que contienen nitrógeno o fósforo.
Un espectrofotómetro infrarrojo por transformada de Fourier (FT—IR) también puede servir como detector. En GC-FT—IR, el efluente de la columna fluye a través de una celda óptica construida a partir de un tubo Pyrex de 10—40 cm con un diámetro interno de 1—3 mm. La superficie interior de la celda está recubierta con una capa reflectante de oro. Las múltiples reflexiones de la radiación fuente a medida que se transmite a través de la célula aumentan la longitud de la trayectoria óptica a través de la muestra. Como es el caso de GC-MS, un detector FT—IR registra continuamente el espectro del eluyente de la columna, lo que nos permite examinar el espectro IR para cualquier incremento de tiempo.