
1.4: Introducción a la Espectroscopia de Absorción Atómica
- Page ID
- 71231

Breve descripción de la espectroscopia de absorción atómica
Historia de la espectroscopia de absorción atómica
La espectroscopia más temprana fue descrita por Marcus Marci von Kronland en 1648 analizando la luz solar a medida que pasa a través de las gotas de agua y creando así un arco iris. Un análisis posterior de la luz solar por William Hyde Wollaston (Figura\(\PageIndex{1}\)) condujo al descubrimiento de líneas negras en el espectro, que en 1820 Sir David Brewster (Figura\(\PageIndex{2}\)) explicó como absorción de la luz en la atmósfera del sol.


Robert Bunsen (Figura\(\PageIndex{3}\)) y Gustav Kirchhoff (Figura\(\PageIndex{4}\)) estudiaron el espectro del sodio y llegaron a la conclusión de que cada elemento tiene su propio espectro único que puede ser utilizado para identificar elementos en la fase vapor. Kirchoff explicó además el fenómeno al afirmar que si un material puede emitir radiación de cierta longitud de onda, también puede absorber radiación de esa longitud de onda. Aunque Bunsen y Kirchoff dieron un gran paso en la definición de la técnica de espectroscopia de absorción atómica (AAS), no fue ampliamente utilizada como técnica analítica excepto en el campo de la astronomía debido a muchas dificultades prácticas.



En 1953, Alan Walsh (Figura\(\PageIndex{5}\)) mejoró drásticamente los métodos de AAS. Abogó por AAS ante muchos fabricantes de instrumentos, pero fue en ningún resultado. A pesar de que había mejorado los métodos, no había demostrado cómo podría ser útil en ninguna aplicación. En 1957, descubrió usos para AAS que convencieron a las manufacturas comercializar los primeros espectrómetros comerciales de AAS. Desde entonces, la popularidad de AAS ha fluctuado a medida que se realizan otras técnicas analíticas y mejoras en los métodos.
Teoría de la espectroscopia de absorción atómica
Para entender cómo funciona la espectroscopia de absorción atómica, es necesaria cierta información de fondo. La teoría atómica comenzó con John Dalton (Figura\(\PageIndex{6}\)) en el siglo XVIII cuando propuso el concepto de átomos, que todos los átomos de un elemento son idénticos, y que los átomos de diferentes elementos pueden combinarse para formar moléculas. En 1913, Niels Bohr (Figura\(\PageIndex{7}\)) revolucionó la teoría atómica al proponer números cuánticos, un núcleo cargado positivamente y electrones que orbitaban alrededor del núcleo en lo que se conoció como el modelo Bohr del átomo. Poco después, Louis DeBroglie (Figura\(\PageIndex{8}\)) propuso energía cuantificada de electrones, que es un concepto extremadamente importante en AAS. Wolfgang Pauli (Figura\(\PageIndex{9}\)) luego elaboró la teoría de DeBroglie al afirmar que no hay dos electrones que puedan compartir los mismos cuatro números cuánticos. Estos descubrimientos históricos en la teoría atómica son necesarios para comprender el mecanismo de AAS.




Los átomos tienen electrones de valencia, que son los electrones más externos del átomo. Los átomos se pueden excitar cuando se irradian, lo que crea un espectro de absorción. Cuando se excita un átomo, el electrón de valencia sube un nivel de energía. Las energías de los diversos estados estacionarios, u órbitas restringidas, pueden entonces ser determinadas por estas líneas de emisión. La línea de resonancia se define entonces como la radiación específica absorbida para alcanzar el estado excitado.
La ecuación de Maxwell-Boltzmann da el número de electrones en cualquier orbital dado. Se relaciona la distribución con la temperatura térmica del sistema (a diferencia de la temperatura electrónica, temperatura vibracional o temperatura rotacional). Plank propuso energía emitida por radiación en paquetes discretos (cuantos),
\[ E= h \nu \nonumber \]
que puede estar relacionado con la ecuación de Einstein
\[ E=mc^2 \label{eq:mc2} \]
Tanto la espectroscopia de emisión atómica como la de absorción atómica pueden ser utilizadas para analizar muestras. La espectroscopia de emisión atómica mide la intensidad de luz emitida por los átomos excitados, mientras que la espectroscopia de absorción atómica mide la luz absorbida por la absorción atómica. Esta luz se encuentra típicamente en la región visible o ultravioleta del espectro electromagnético. Luego se compara el porcentaje con una curva de calibración para determinar la cantidad de material en la muestra. La energía del sistema puede ser utilizada para encontrar la frecuencia de la radiación, y así la longitud de onda a través de la combinación de ecuaciones\ ref {eq:mc2} y\ ref {eq:ncl}.
\[ \nu = c/\lambda \label{eq:ncl} \]
Debido a que se cuantifican los niveles de energía, solo se permiten ciertas longitudes de onda y cada átomo tiene un espectro único. Existen muchas variables que pueden afectar al sistema. Por ejemplo, si la muestra se cambia de manera que aumente la población de átomos, habrá un incremento tanto en la emisión como en la absorción y viceversa. También hay variables que afectan la relación de átomos excitados a no excitados como un aumento en la temperatura del vapor.
Aplicaciones de la espectroscopia de absorción atómica
Existen muchas aplicaciones de la espectroscopia de absorción atómica (AAS) debido a su especificidad. Estos se pueden dividir en las amplias categorías de análisis biológico, análisis ambiental y marino, y análisis geológico.
Análisis biológico
Las muestras biológicas pueden incluir tanto muestras de tejido humano como muestras de alimentos. En muestras de tejido humano, el AAS se puede utilizar para determinar la cantidad de diversos niveles de metales y otros electrolitos, dentro de las muestras de tejido. Estas muestras de tejido pueden ser muchas cosas, incluyendo pero no limitado a sangre, médula ósea, orina, cabello y uñas. La preparación de la muestra depende de la muestra. Esto es extremadamente importante ya que muchos elementos son tóxicos en ciertas concentraciones en el cuerpo, y AAS puede analizar en qué concentraciones están presentes. Algunos ejemplos de oligoelementos para los que se analizan las muestras son arsénico, mercurio y plomo.
Un ejemplo de aplicación de AAS a tejido humano es la medición de los electrolitos de sodio y potasio en plasma. Esta medición es importante porque los valores pueden ser indicativos de diversas enfermedades cuando están fuera del rango normal. El método típico utilizado para este análisis es la atomización de una dilución 1:50 en cloruro de estroncio (\(\ce{SrCl2}\)) usando una llama aire-hidrógeno. El sodio se detecta en su línea secundaria (330.2 nm) porque la detección en la primera línea requeriría dilución adicional de la muestra debido a la intensidad de la señal. La razón por la que se utiliza cloruro de estroncio es porque reduce la ionización de los iones potasio y sodio, al tiempo que elimina la interferencia de fosfato y calcio.
En la industria alimentaria, AAS proporciona análisis de verduras, productos animales y alimentos para animales. Este tipo de análisis son algunas de las aplicaciones más antiguas de AAS. Una consideración importante que debe tenerse en cuenta en el análisis de alimentos es el muestreo. La muestra debe ser una representación precisa de lo que se está analizando. Debido a esto, debe ser homogéneo, y muchas veces se necesita que se procesen varias muestras. La mayoría de las veces se realizan muestras de alimentos para determinar las cantidades de minerales y oligoelementos para que los consumidores sepan si están consumiendo una cantidad adecuada. También se analizan muestras para determinar metales pesados que pueden ser perjudiciales para los consumidores.
Análisis ambiental y marino
El análisis ambiental y marino generalmente se refiere al análisis de agua de varios tipos. El análisis del agua incluye muchas cosas que van desde agua potable hasta aguas residuales y agua de mar. A diferencia de las muestras biológicas, la preparación de muestras de agua se rige más por las leyes que por la propia muestra. Los analitos que se pueden medir también varían mucho y a menudo pueden incluir plomo, cobre, níquel y mercurio.
Un ejemplo de análisis de agua es un análisis de lixiviación de plomo y zinc de soldadura de estaño-plomo en agua. La soldadura es lo que une las juntas de los tubos de cobre. En este experimento en particular, se analizaron agua blanda, agua ácida y agua clorada. La preparación de la muestra consistió en exponer las diversas muestras de agua a placas de cobre con soldadura durante varios intervalos de tiempo. Luego se analizaron las muestras para detectar cobre y zinc con llama aire-acetileno AAS. Se utilizó una lámpara de deuterio. Para las muestras que tenían niveles de cobre por debajo de 100 µg/L, el método se cambió a AAS electrotérmico de horno de grafito debido a su mayor sensibilidad.
Análisis geológico
El análisis geológico abarca tanto las reservas minerales como la investigación ambiental. Al prospeccionar reservas minerales, el método de AAS utilizado necesita ser barato, rápido y versátil porque la mayoría de las perspectivas terminan siendo de ningún uso económico. Al estudiar rocas, la preparación puede incluir digestiones ácidas o lixiviación. Si la muestra necesita analizar el contenido de silicio, la digestión ácida no es un método de preparación adecuado.
Un ejemplo es el análisis de sedimentos lacustres y fluviales para detectar plomo y cadmio. Debido a que este experimento involucra una muestra sólida, se necesita más preparación que para los otros ejemplos. El sedimento primero se secó, luego se trituró en polvo, y luego se descompuso en una bomba con ácido nítrico (\(\ce{HNO3}\)) y ácido perclórico (\(\ce{HClO4}\)). Se prepararon estándares de plomo y cadmio. Se añadieron sulfato de amonio (\(\ce{[NH4][SO4]}\)\(\ce{[NH4][3PO4]}\)]) y fosfato amónico (]) a las muestras para corregir las interferencias causadas por sodio y potasio que están presentes en la muestra. Los estándares y muestras se analizaron con AAS electrotérmico.
Instrumentación
Atomizador
Para que la muestra sea analizada, primero debe ser atomizada. Este es un paso sumamente importante en AAS porque determina la sensibilidad de la lectura. Los atomizadores más efectivos crean una gran cantidad de átomos libres homogéneos. Existen muchos tipos de atomizadores, pero solo se utilizan comúnmente dos: atomizadores de llama y electrotérmicos.
Atomizador de llama
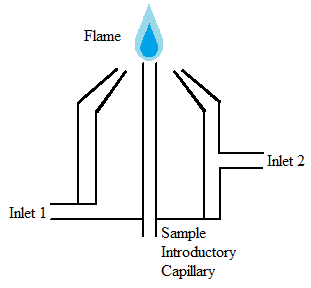
Los atomizadores de llama (Figura\(\PageIndex{10}\)) son ampliamente utilizados por una multitud de razones, incluyendo su simplicidad, bajo costo y largo período de tiempo que han sido utilizados. Los atomizadores de llama aceptan un aerosol de un nebulizador a una llama que tiene suficiente energía para volatilizar y atomizar la muestra. Cuando esto sucede, la muestra se seca, vaporiza, atomiza e ioniza. Dentro de esta categoría de atomizadores, existen muchas subcategorías determinadas por la composición química de la llama. La composición de la llama a menudo se determina en base a la muestra que se analiza. La llama en sí debe cumplir con varios requisitos, incluida la energía suficiente, una longitud larga, no turbulenta y segura.
Atomizador electrotérmico
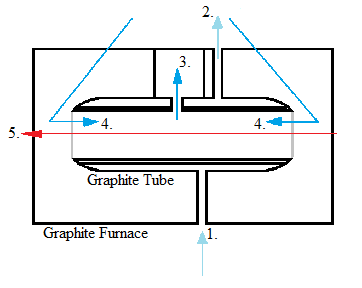
Aunque los atomizadores electrotérmicos se desarrollaron antes que los atomizadores de llama, no se hicieron populares hasta más recientemente debido a las mejoras realizadas en el nivel de detección. Emplean tubos de grafito que aumentan la temperatura de manera escalonada. La atomización electrotérmica primero seca la muestra y evapora gran parte del disolvente y las impurezas, luego atomiza la muestra y luego la eleva a una temperatura extremadamente alta para limpiar el tubo de grafito. Algunos requisitos para esta forma de atomización son la capacidad de mantener una temperatura constante durante la atomización, tener una atomización rápida, mantener un gran volumen de solución y emitir radiación mínima. La atomización electrotérmica es mucho menos dura que el método de atomización por llama.
Fuente de radiación
La fuente de radiación irradia entonces la muestra atomizada. La muestra absorbe parte de la radiación, y el resto pasa a través del espectrómetro a un detector. Las fuentes de radiación se pueden separar en dos amplias categorías: fuentes lineales y fuentes continuas. Las fuentes de línea excitan el analito y así emiten su propio espectro lineal. Las lámparas de cátodo hueco y las lámparas de descarga sin electrodos son los ejemplos más utilizados de fuentes de línea. Por otro lado, las fuentes continuas tienen radiación que se extiende a lo largo de un rango más amplio de longitudes de onda. Por lo general, estas fuentes solo se utilizan para la corrección de fondo. Las lámparas de deuterio y las lámparas halógenas se utilizan a menudo para este propósito.
Espectrómetro
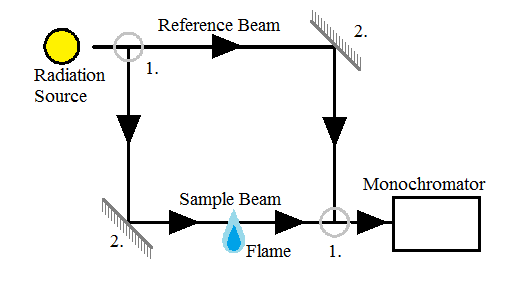
Los espectrómetros se utilizan para separar las diferentes longitudes de onda de la luz antes de que pasen al detector. El espectrómetro utilizado en AAS puede ser de un solo haz o de doble haz. Los espectrómetros de haz único solo requieren radiación que pase directamente a través de la muestra atomizada, mientras que los espectrómetros de doble haz (Figura\(\PageIndex{12}\)), como implica el nombre, requieren dos haces de luz; uno que pasa directamente por la muestra, y otro que no pasa por la muestra en absoluto. (Insertar diagramas) Los espectrómetros de un solo haz tienen menos componentes ópticos y, por lo tanto, sufren menos pérdida de radiación. Los monocromadores de doble haz tienen más componentes ópticos, pero también son más estables con el tiempo porque pueden compensar los cambios más fácilmente.
Obtención de Mediciones
Preparación de muestras
La preparación de las muestras es extremadamente variada debido al rango de muestras que se pueden analizar. Independientemente del tipo de muestra, se deben hacer ciertas consideraciones. Estos incluyen el ambiente de laboratorio, el recipiente que contiene la muestra, el almacenamiento de la muestra y el pretratamiento de la muestra.
La preparación de las muestras comienza con tener un ambiente limpio para trabajar. A menudo se usa AAS para medir oligoelementos, en cuyo caso la contaminación puede conducir a un error grave. El equipo posible incluye campanas de flujo laminar, salas limpias y recipientes cerrados y limpios para el transporte de la muestra. No solo se debe mantener limpia la muestra, sino que también debe conservarse en términos de pH, constituyentes y cualquier otra propiedad que pueda alterar el contenido.
Cuando se almacenan oligoelementos, el material de las paredes de los vasos puede adsorber parte del analito, lo que lleva a malos resultados. Para corregir esto, a menudo se utilizan polímeros de perfluoroalcoxi (PFA), sílice, carbono vítreo y otros materiales con superficies inertes como material de almacenamiento. La acidificación de la solución con ácido clorhídrico o nítrico también puede ayudar a evitar que los iones se adhieran a las paredes del recipiente al competir por el espacio. Los recipientes también deben contener una superficie mínima para minimizar los posibles sitios de adsorción.
El pretratamiento de la muestra depende de la naturaleza de la muestra. Consulte la Tabla\(\PageIndex{1}\) para conocer los métodos de pretratamiento de muestras.
| Muestra | Ejemplos | Método de pretratamiento |
|---|---|---|
| Soluciones acuosas | Agua, bebidas, orina, sangre | Digestión si están presentes sustituyentes causantes de interferencia |
| Suspensiones | Agua, bebidas, orina, sangre | La materia sólida debe eliminarse por filtración, centrifugación o digestión, y luego se pueden seguir los métodos para soluciones acuosas |
| Líquidos orgánicos | Combustibles, aceites | Ya sea medición directa con AAS o dilatación con material orgánico seguido de medición con AAS, los estándares deben contener el analito en la misma forma que la muestra |
| Sólidos | Alimentos, rocas | Digestión seguida de AAS electrotérmico |
Curva de calibración
Para determinar la concentración del analito en la solución, se pueden emplear curvas de calibración. Usando estándares, se puede crear una gráfica de concentración versus absorbancia. Tres métodos comunes utilizados para hacer curvas de calibración son la técnica de calibración estándar, la técnica de horquillado y la técnica de adición de analitos.
Técnica de calibración estándar
Esta técnica es tanto la más simple como la más utilizada. La concentración de la muestra se encuentra comparando su absorbancia o absorbancia integrada con una curva de la concentración de los patrones frente a las absorbancias o absorbancias integradas de los estándares. Para que se aplique este método se deben cumplir las siguientes condiciones:
- Tanto los estándares como la muestra deben tener el mismo comportamiento cuando se atomizan. Si no lo hacen, la matriz de los estándares debe ser alterada para que coincida con la de la muestra.
- El error en la medición de la absorbancia debe ser menor que el de la preparación de los estándares.
- Las muestras deben ser homogéneas.
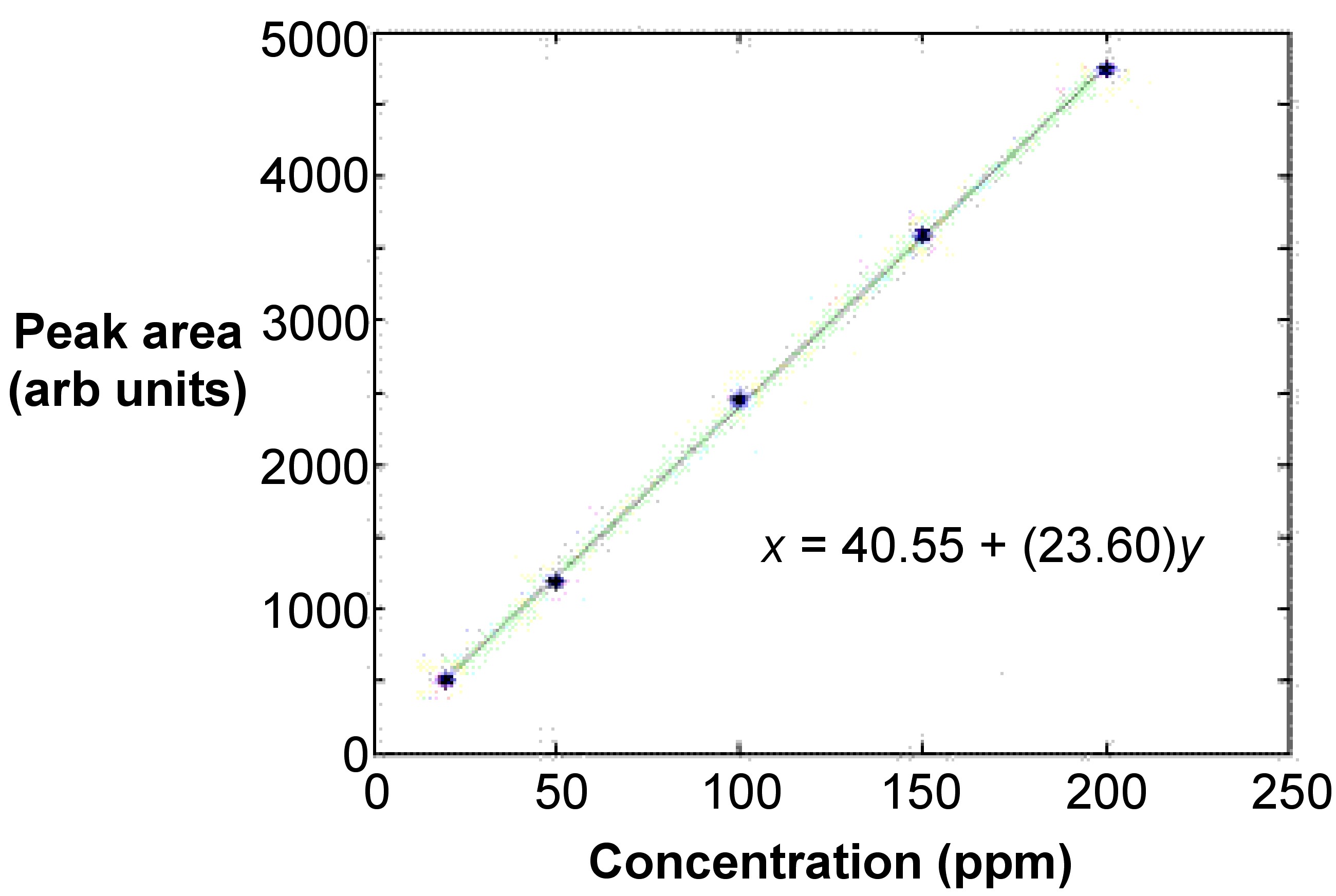
La curva es típicamente lineal e involucra al menos cinco puntos de cinco patrones que están a concentraciones equidistantes entre sí (Figura\(\PageIndex{13}\)). Esto asegura que el ajuste sea aceptable. Se utiliza un cálculo de mínimos cuadrados medios para ajustar linealmente la línea. En la mayoría de los casos, la curva es lineal solo hasta valores de absorbancia de 0.5 a 0.8. Los valores de absorbancia de los estándares deben tener restado el valor de absorbancia de un blanco.
Técnica de Horquillado
La técnica de horquillado es una variación de la técnica de calibración estándar. En este método, solo son necesarios dos estándares con concentraciones\(c_1\) y\(c_2\). Encuadernan muy de cerca el valor aproximado de la concentración de la muestra. Aplicando la Ecuación\ ref {horquillado} para determinar el valor para la muestra, donde\(c_x\) y\(A_x\) son la concentración y adsorbancia de lo desconocido,\(A_1\) y y\(A_2\) son la adsorbancia para\(c_1\) y\(c_2\), respectivamente.
\[ c _ { x } = \frac { \left( A _ { x } - A _ { 1 } \right) \left( c _ { 1 } - c _ { 2 } \right) } { A _ { 2 } - A _ { 1 } } + c _ { 1 } \label{bracketing } \]
Este método es muy útil cuando la concentración del analito en la muestra está fuera de la porción lineal de la curva de calibración porque el corchete es tan pequeño que la porción de la curva que se está utilizando puede ser retratada como lineal. Aunque este método se puede utilizar con precisión para curvas no lineales, cuanto más lejos esté la curva de lineal mayor será el error. Para ayudar a reducir este error, los estándares deben cortejar la muestra muy de cerca.
Técnica de adición de analitos
La técnica de adición de analito se utiliza a menudo cuando se espera que los concomitantes en la muestra creen muchas interferencias y se desconoce la composición de la muestra. Las dos técnicas anteriores requieren que los estándares tengan una matriz similar a la de la muestra, pero eso no es posible cuando se desconoce la matriz. Para compensar esto, la técnica de adición de analitos utiliza una alícuota de la muestra misma como matriz. A continuación, las alícuotas se adicionan con diversas cantidades del analito. Esta técnica debe ser utilizada sólo dentro del rango lineal de las absorbancias.
Interferencia de medición
La interferencia es causada por contaminantes dentro de la muestra que absorben a la misma longitud de onda que el analito, y por lo tanto pueden causar mediciones inexactas. Las correcciones se pueden hacer a través de una variedad de métodos tales como corrección de fondo, adición de aditivos químicos o adición de analito.
| Tipo de interferencia | Causa de interferencia | Resultado | Ejemplo | Medidas de corrección |
|---|---|---|---|---|
| Superposición de línea atómica | El perfil espectral de dos elementos está dentro de 0.01 nm entre sí | Mayor valor de absorción experimental que el valor real | Muy raro, siendo el único problema plausable el del cobre (324,754 nm) y el europio (324,753 nm) | Normalmente no ocurre en situaciones prácticas, por lo que no existe un método de corrección establecido |
| Solapamiento molecular de bandas y líneas | El perfil espectral de un elemento se solapa con la banda molecular | Mayor valor de absorción experimental que el valor real | Hidróxido de calcio y bario a 553.6 nm en una llama de aire-acetileno | Corrección de fondo |
| Ionización (fase vapor o potenciación catiónica) | los átomos se ionizan a la temperatura de la llama/horno, lo que disminuye la cantidad de átomos libres | Menor valor de absorción experimental que el valor real | Los problemas ocurren comúnmente con cesio, potasio y sodio | Añadir un supresor de ionización (o tampón) tanto a la muestra como a los estándares |
| Dispersión de luz | Las partículas sólidas dispersan el haz de luz disminuyendo la intensidad del haz que entra en el monocromador | Mayor valor de absorción experimental que el valor real | Alto en muestras con muchos elementos refractarios, más alto en longitudes de onda UV (agregue un ejemplo específico) | Modificación de matriz y/o corrección de fondo |
| Químico | La sustancia química que se analiza está contenida dentro de un compuesto en el analito que no está atomizado | Menor valor de absorción experimental que el valor real | Los iones de calcio y fosfato forman fosfato de calcio que luego se convierte en pirofosfato de calcio que es estable a altas temperaturas | Aumente la temperatura de la llama si se usa AAS de llama, use un producto químico de liberación o adición estándar para AAS electrotérmico |
| Físico | Si las propiedades físicas de la muestra y los patrones son diferentes, la atomización puede verse afectada afectando así el número de población de átomos libres | Puede variar en cualquier dirección dependiendo de las condiciones | Diferencias de viscosidad, diferencias de tensión superficial, etc. | Alterar los estándares para que tengan propiedades físicas similares a las de las muestras |
| Volitalización | En la atomización electrotérmica, se producirá interferencia si la tasa de volatilización no es la misma para la muestra que para el estándar, lo que a menudo es causado por una matriz pesada | Puede variar en cualquier dirección dependiendo de las condiciones | Los cloruros son muy volátiles, por lo que necesitan ser convertidos a una forma menos volátil. A menudo esto se hace mediante la adición de nitrato o slufate. El zinc y el plomo también son altamente problemáticos | Cambiar la matriz por adición estándar, o volatilizar selectivamente los componentes de la matriz |
Bibliografía
- L. Ebon, A. Fisher y S. J. Hill, Una introducción a la espectrometría atómica analítica, Ed. E. H. Evans, Wiley, Nueva York (1998).
- B. Welz y M. Sperling, Espectrometría de Absorción Atómica, 3 rd Ed, Wiley-VCH, Nueva York (1999).
- J. W. Robinson, Espectroscopia Atómica, 2ª Ed. Marcel Dekker, Inc., Nueva York (1996).
- K. S. Subramanian, Water Res., 1995, 29, 1827.
- M. Sakata y O. Shimoda, Water Res., 1982, 16, 231.
- J. C. Van Loon, Espectroscopia Analítica de Absorción Atómica Métodos Seleccionados, Academic Press, Nueva York (1980).