2.7: Electroquímica
- Page ID
- 71107

Mediciones de voltamperometría cíclica
Introducción
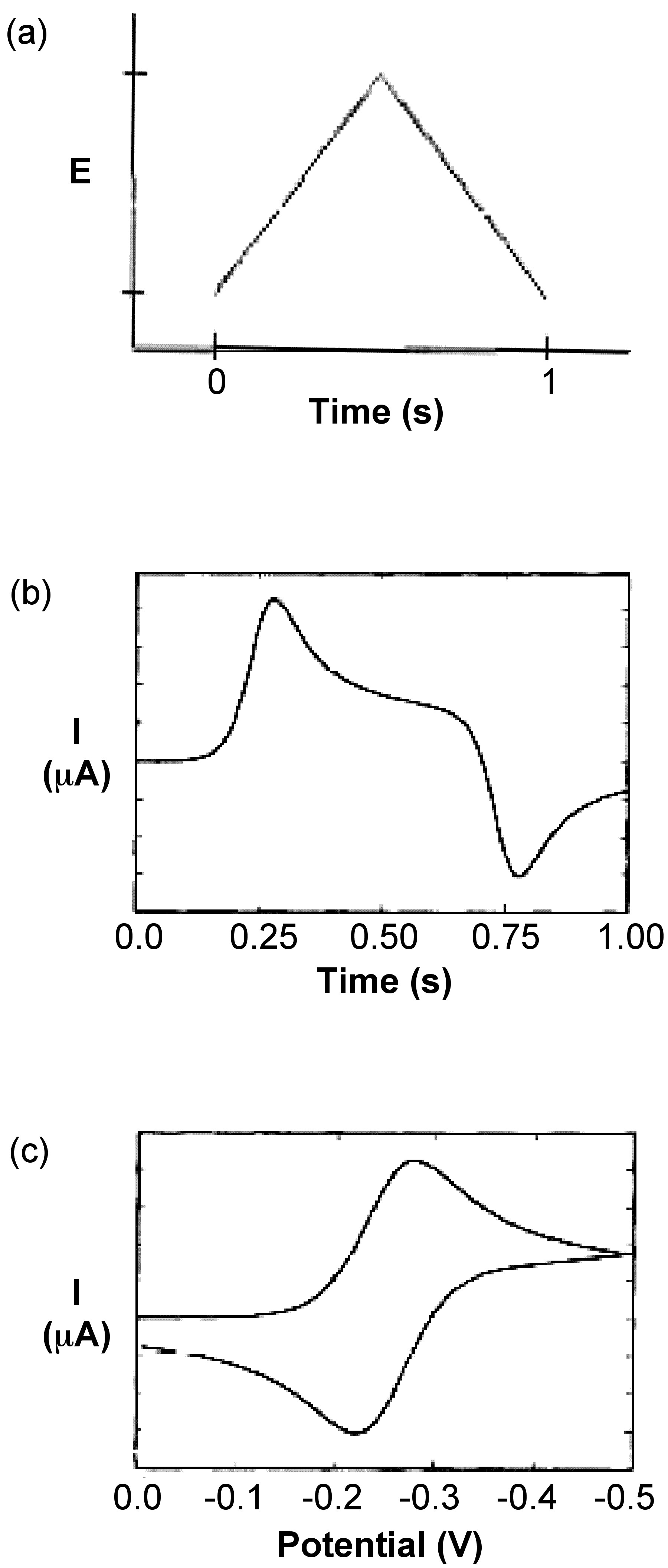
La voltamperometría cíclica (CV) es un tipo de mediciones electroquímicas potenciodinámicas. En términos generales, el proceso de operación es un experimento reversible controlado por potencial, que escanea el potencial eléctrico antes de girar a la dirección inversa después de alcanzar el potencial final y luego escanea de nuevo al potencial inicial, como se muestra en la Figura\(\PageIndex{1}\) -a. Cuando se aplica voltaje al sistema cambia con el tiempo, la corriente cambiará con el tiempo en consecuencia como se muestra en la Figura\(\PageIndex{1}\) -b. Así, la curva de corriente y voltaje, ilustrada en la Figura\(\PageIndex{1}\) -c, se puede representar a partir de los datos, los cuales se pueden obtener de la Figura\(\PageIndex{1}\) -a y la Figura\(\PageIndex{1}\) -b.
La voltamperometría cíclica es una caracterización analítica muy importante en el campo de la electroquímica. Cualquier proceso que incluya transferencia de electrones puede ser investigado con esta caracterización. Por ejemplo, la investigación de reacciones catalíticas, el análisis de la estequiometría de compuestos complejos y la determinación de la banda prohibida de los materiales fotovoltaicos. En este módulo, me enfocaré en la aplicación de la medición de CV en el campo de la caracterización de materiales de células solares.
Aunque el CV se practicó por primera vez usando un electrodo de gota de mercurio colgante, basado en el trabajo del ganador del Premio Nobel Heyrovský (Figura\(\PageIndex{2}\)), no se generalizó hasta que se utilizaron electrodos sólidos como Pt, Au y electrodos carbonosos, particularmente para estudiar las oxidaciones anódicas. Se hizo un gran avance cuando se dieron a conocer los diagnósticos mecanicistas y las cuantificaciones acompañantes a través de las simulaciones por computadora. Ahora, la aplicación de computadoras y paquetes de software relacionados hacen que el análisis de datos sea mucho más rápido y fácil.

Los componentes de un sistema CV
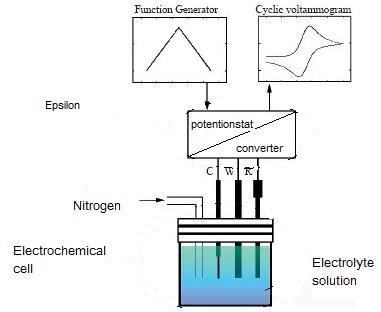
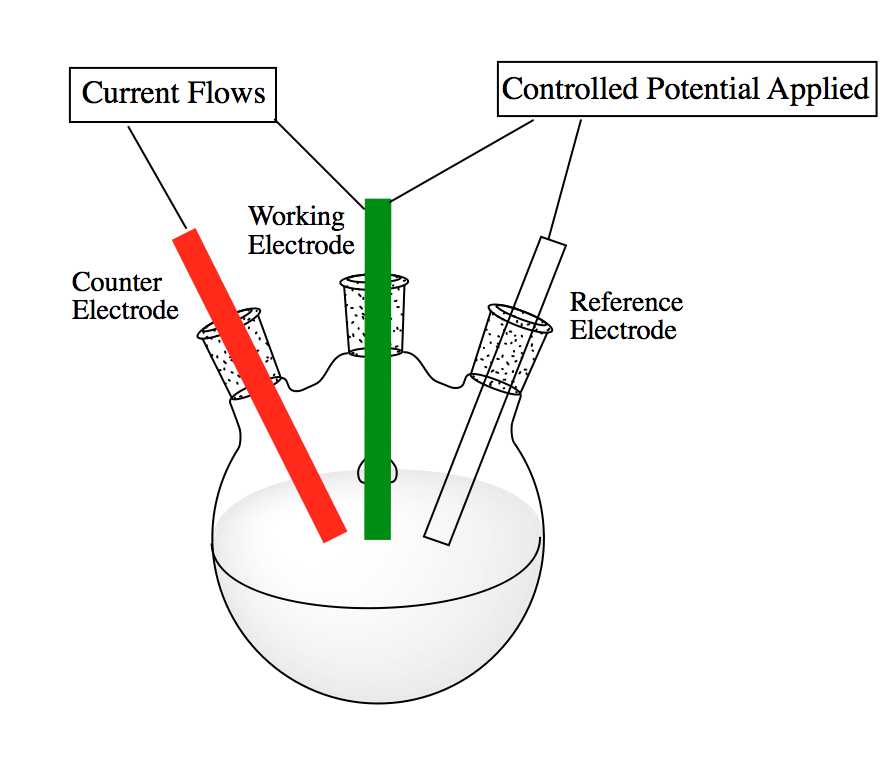
Como se muestra en la Figura\(\PageIndex{3}\), los sistemas CV son los siguientes:
- El épsilon incluye potenciostato y convertidor de corriente-voltaje. El potenciostato es necesario para controlar el potencial aplicado, y se utiliza un convertidor de corriente a voltaje para medir la corriente, ambos contenidos dentro del épsilon (Figura\(\PageIndex{3}\).
- El sistema de entrada es un generador de funciones (Figura\(\PageIndex{3}\). Los operadores pueden cambiar parámetros, incluyendo la velocidad de escaneo y el rango de escaneo, a través de esta parte. La parte de salida es una pantalla de computadora, que puede mostrar datos y curvas directamente a los operadores.
- Todos los electrodos deben funcionar en solución electrolítica.
- En ocasiones, el oxígeno y el agua en la atmósfera se disolverán en la solución, y se desoxidarán u oxidarán cuando se aplique voltaje. Por lo tanto, los datos serán menos precisos. Para evitar que esto suceda, se requiere burbujear un gas inerte (nitrógeno o argón).
- El componente clave de los sistemas CV es la celda electroquímica que está conectada a la parte épsilon. La celda electroquímica contiene tres electrodos, contraelectrodo (C en la Figura\(\PageIndex{3}\)) electrodo de trabajo (W en la Figura\(\PageIndex{3}\)) y electrodo de referencia (R en la Figura\(\PageIndex{3}\)). Todos ellos deben estar sumergidos en una solución electrolítica cuando se trabaja.
Para comprender mejor los electrodos mencionados anteriormente, se discutirán con más detalle tres tipos de electrodos.
- Los contraelectrodos (C en la Figura\(\PageIndex{3}\) son electrodos de alta área superficial no reactivos, para los cuales la gasa de platino es la elección común.
- El electrodo de trabajo en (W en la Figura\(\PageIndex{3}\)) es comúnmente un disco con incrustaciones de electrodos (Pt, Au, grafito, etc.) de área bien definida son los más utilizados. Otras geometrías pueden estar disponibles en circunstancias apropiadas, como caída o suspensión del hemisferio de mercurio, cilindro, banda, matrices y electrodos de rejilla.
- Para el electrodo de referencia (R en la Figura\(\PageIndex{3}\)) se utilizan comúnmente medias celdas acuosas de Ag/AgCl o calomelanos, y se pueden obtener comercial o fácilmente preparadas en el laboratorio. En ocasiones, se utiliza un simple alambre de plata o platino junto con una referencia de potencial interno proporcionada por el ferroceno, cuando no se dispone de un electrodo de referencia convencional adecuado. El ferroceno sufre una oxidación de un electrón a un potencial bajo, alrededor de 0.5 V versus un electrodo de calomelano saturado (SCE). También se ha utilizado como estándar en electroquímica como F c + /F c = 0.64 V versus un electrodo de hidrógeno normal (NHE).
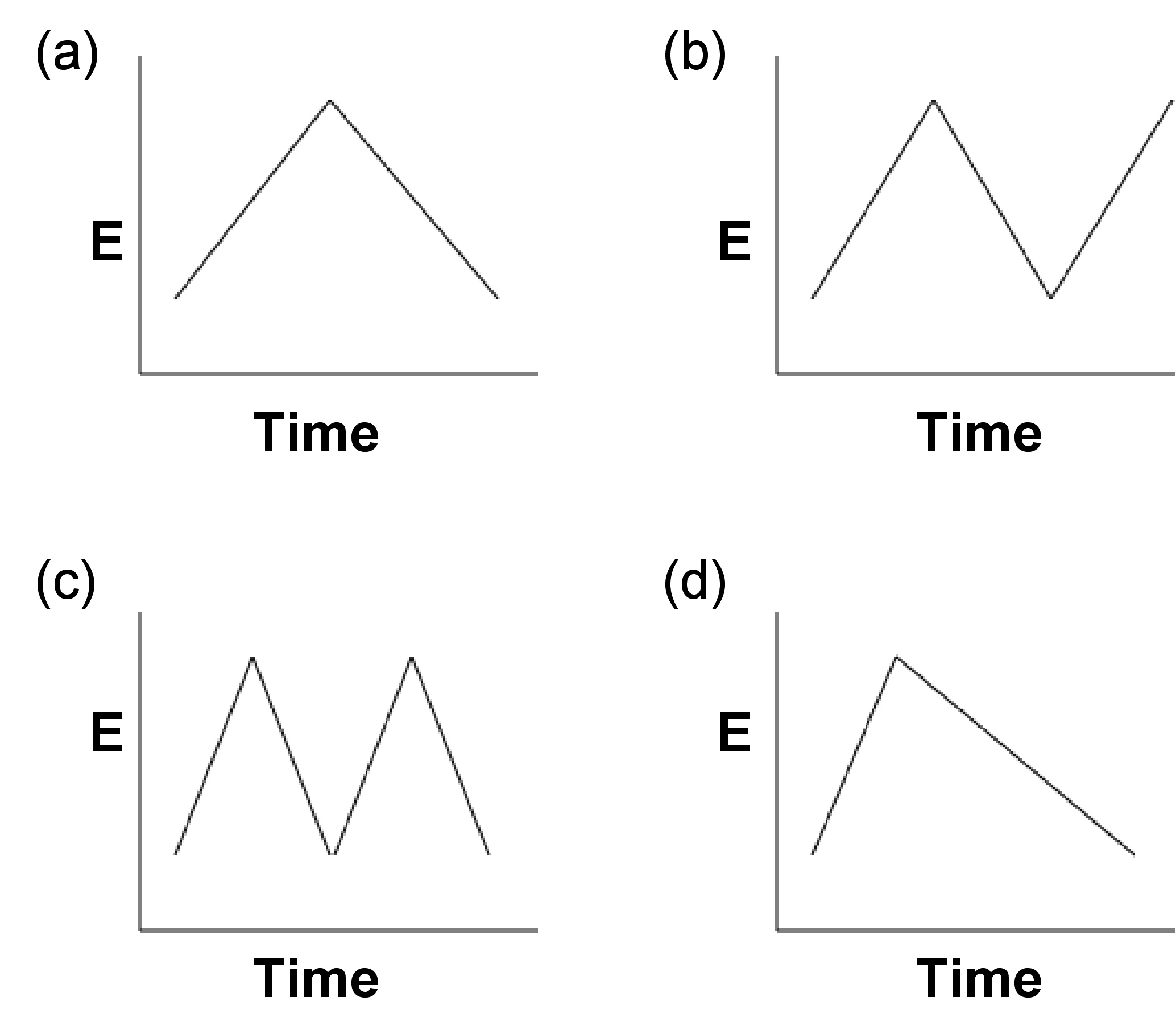
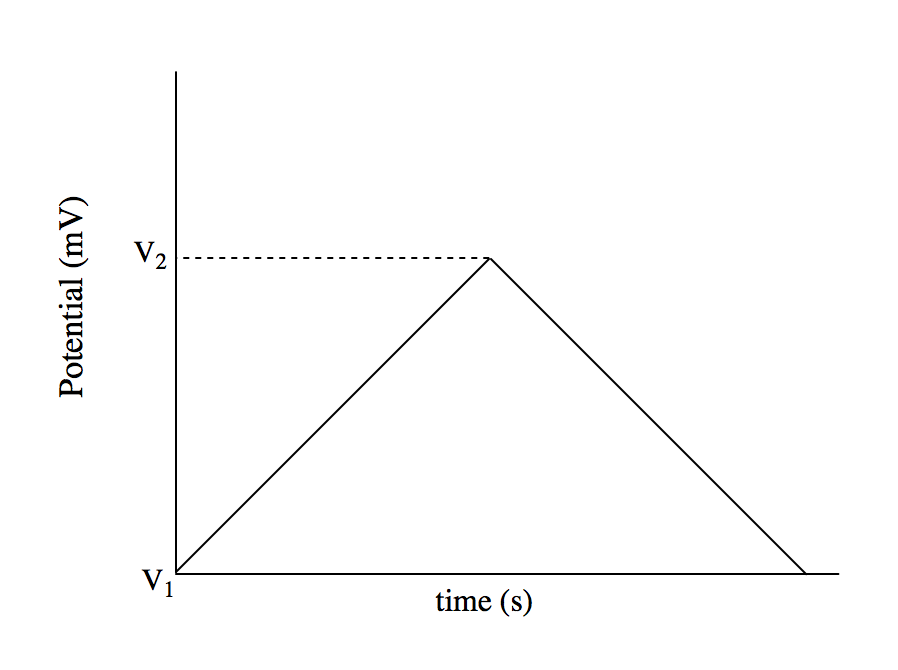
Los sistemas de voltamperometría cíclica emplean diferentes tipos de formas de onda potenciales (Figura\(\PageIndex{4}\)) que pueden usarse para satisfacer diferentes requisitos. Las formas de onda potenciales reflejan la forma en que se aplica el potencial a este sistema. Estos diferentes tipos son referidos por nombres característicos, por ejemplo, voltamperometría cíclica y voltamperometría diferencial de pulso. El método analítico de voltametría cíclica es aquel cuya forma de onda potencial es generalmente un triángulo isósceles (Figura\(\PageIndex{4}\) a).
Principios Físicos de los Sistemas CV
Como se mencionó anteriormente, hay dos partes principales de un sistema CV: la celda electroquímica y la épsilon. La figura\(\PageIndex{6}\) muestra el dibujo esquemático del diagrama de circuito en celda electroquímica.
En un experimento voltamétrico, se aplica potencial a un sistema, utilizando el electrodo de trabajo (W en la Figura\(\PageIndex{7}\)) y el electrodo de referencia (R = Figura\(\PageIndex{7}\)) y la respuesta de corriente se mide usando el electrodo de trabajo y un tercer electrodo, el contraelectrodo (C en la Figura\(\PageIndex{7}\)). La curva típica de corriente-voltaje para ferricianuro/ferrocianuro,\ ref {1}, se muestra en la Figura\(\PageIndex{7}\).
\[ E_{eq} \ =\ E^{\circ ' } \ +\ ( 0.059/n ) \ log( [ reactant ] / [ product ] ) \label{1} \]
Qué información útil podemos obtener de los datos recopilados
La información que podemos obtener de los datos experimentales CV es la curva corriente-voltaje. A partir de la curva podemos entonces determinar el potencial redox, y obtener conocimientos sobre la cinética de las reacciones electrónicas, así como determinar la presencia de intermedio de reacción.
Por qué CV para las caracterizaciones de materiales de células solares
A pesar de algunas limitaciones, la voltametría cíclica es muy adecuada para una amplia gama de aplicaciones. Además, en algunas áreas de investigación, la voltametría cíclica es una de las técnicas estándar utilizadas para la caracterización. Debido a sus características formas de curvas, ha sido considerada como 'espectroscopía electroquímica'. Además, el sistema es bastante fácil de operar, y la preparación de la muestra es relativamente simple.
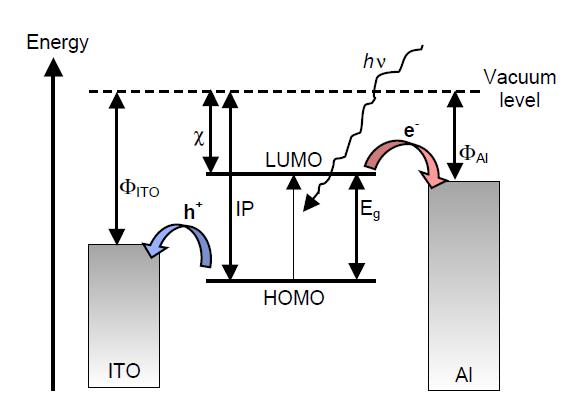
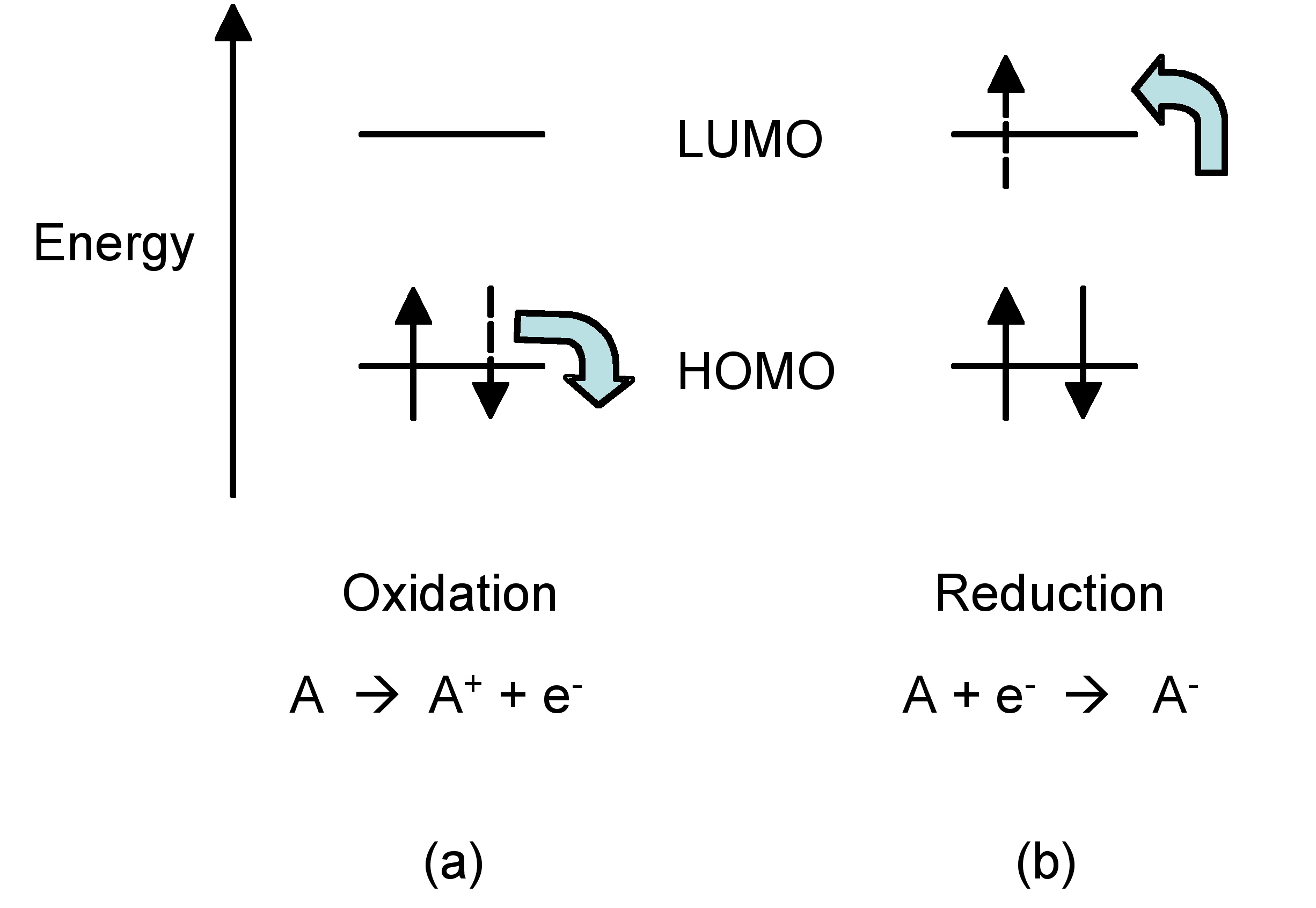
El intervalo de banda de un semiconductor es un valor muy importante a determinar para materiales fotovoltaicos. La figura\(\PageIndex{8}\) muestra el nivel de energía relativo involucrado en la recolección de luz de una célula solar orgánica. La diferencia de energía (E g) entre el orbital molecular desocupado más bajo (LUMO) y el orbital molecular ocupado más alto (HOMO), lo que determina la eficiencia. La oxidación y reducción de una molécula orgánica implica transferencias de electrones (Figura\(\PageIndex{9}\)), y las mediciones de CV pueden ser utilizadas para determinar el cambio potencial durante el redox. Mediante el análisis de los datos obtenidos por la medición de CV se obtiene la brecha de banda electrónica.
El Ejemplo Del Análisis De Datos CV En Carecterización De Material De Células Solares
Los nanobones de grafeno (GNR) son láminas largas y estrechas de grafeno formadas a partir de la descompresión de nanotubos de carbono (Figura\(\PageIndex{10}\)). Los GNR pueden ser tanto semiconductores como semimetálicos, dependiendo de su anchura, y representan una variedad particularmente versátil de grafeno. La alta superficie, la alta relación de aspecto y las interesantes propiedades electrónicas de los GNR los convierten en candidatos prometedores para aplicaciones de materiales de almacenamiento de energía.

Los nanobones de grafeno se pueden oxidar a nanorobones de grafeno oxidados (XGNR), son fácilmente solubles en agua fácilmente. La voltametría cíclica es un método eficaz para caracterizar la banda prohibida de materiales semiconductores. Para probar la brecha de banda de nanorobones de grafeno oxidado (XGNR), los parámetros operativos se pueden establecer de la siguiente manera:
- Solución KCl 0.1M
- Electrodo de trabajo: oro evaporado sobre silicio.
- Velocidad de escaneo: 10 Mv/s.
- Rango de barrido: 0 ~ 3000 mV para reacción de oxidación; -3000 ~ 0 mV para reacción de reducción.
- Preparación de las muestras: aplicar por centrifugación una solución acuosa de los nanorobones de grafeno oxidado sobre el electrodo de trabajo y secar a 100 °C.
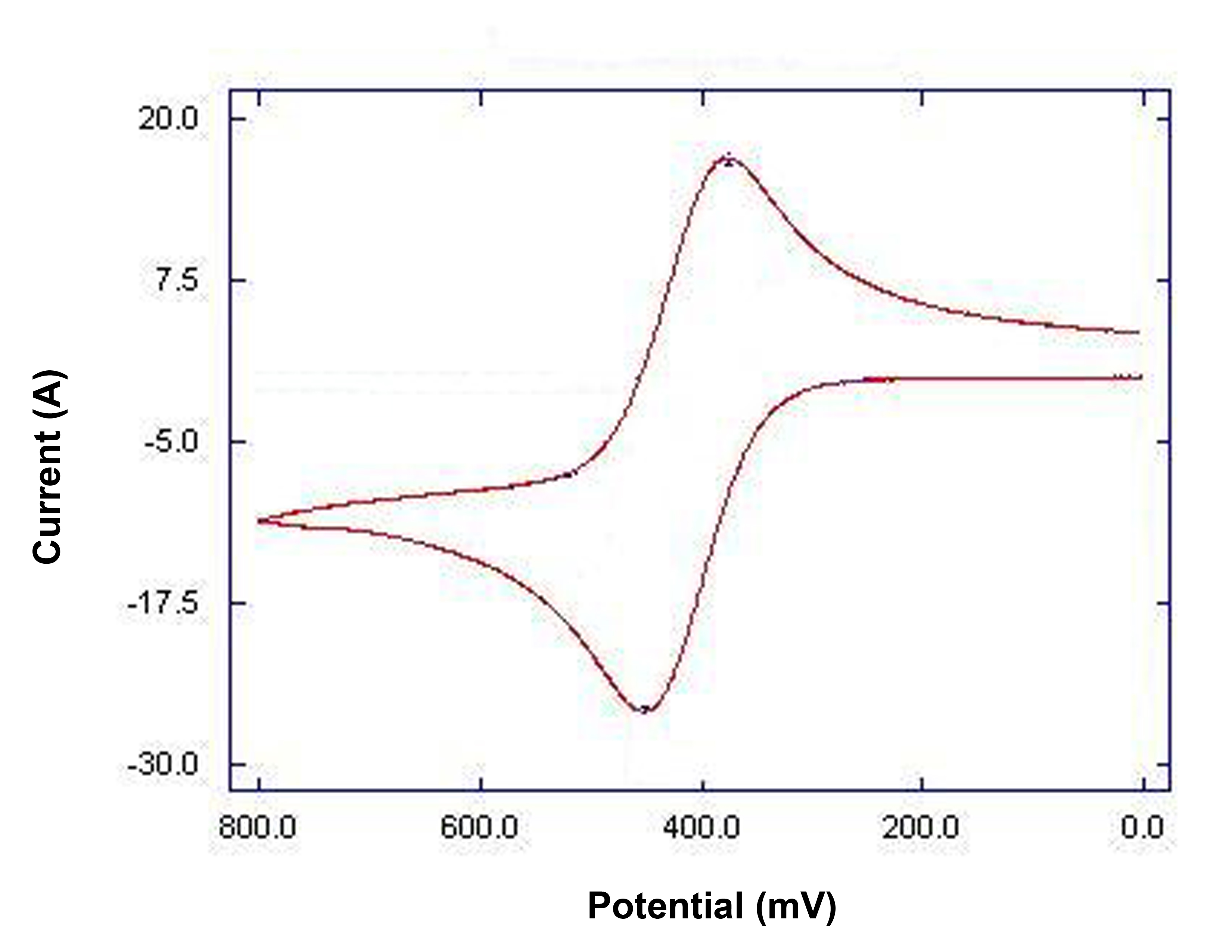
Para asegurarse de que los resultados son precisos, se pueden probar dos muestras bajo las mismas condiciones para ver si los picos redox están en la misma posición. La cantidad de XGNR variará de una muestra a otra, por lo que la altura de los picos también variará. Las curvas típicas obtenidas de la reacción de oxidación (Figura\(\PageIndex{9}\) a) y la reacción de reducción (Figura\(\PageIndex{9}\) b) se muestran en la Figura\(\PageIndex{10}\) y Figura\(\PageIndex{11}\), respectivamente.
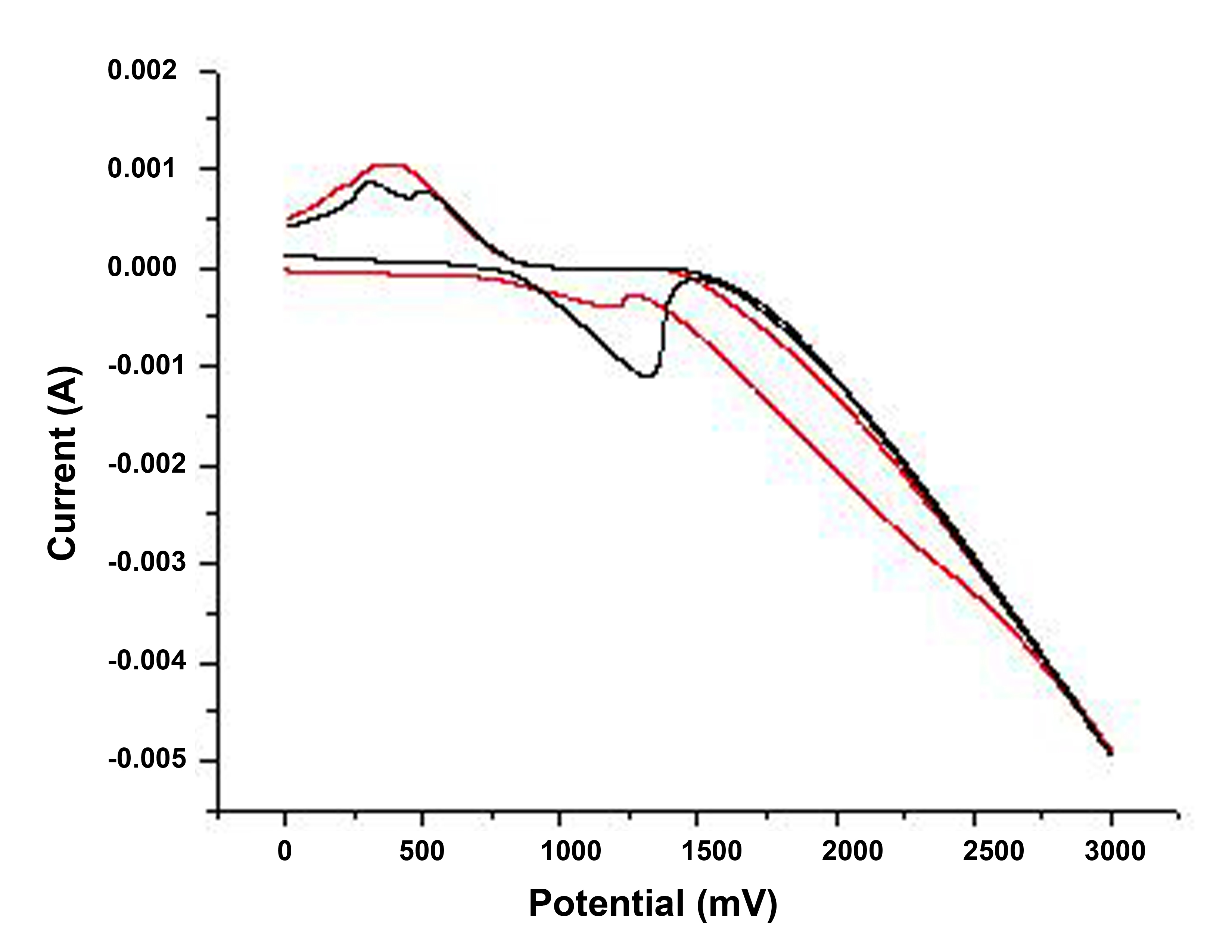
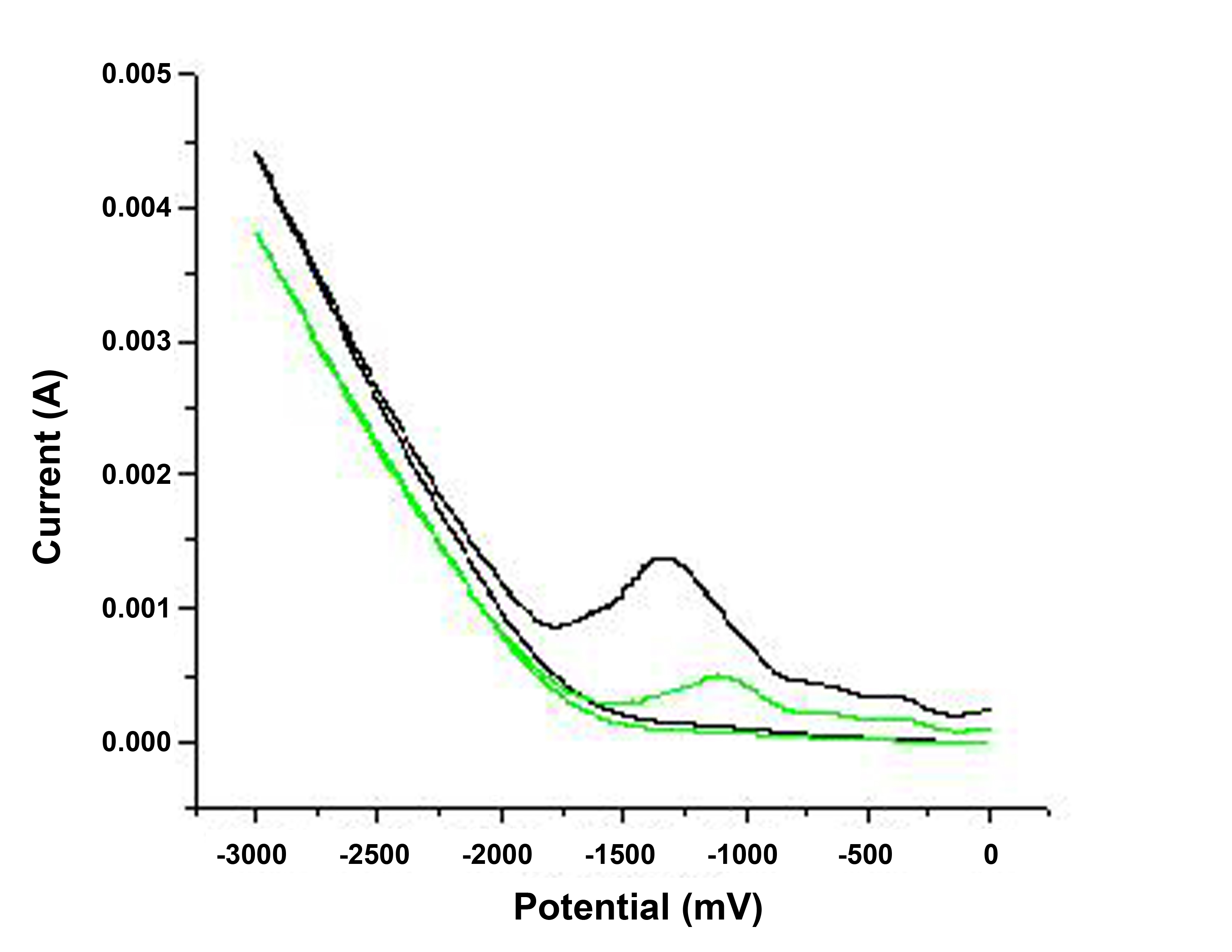
De las curvas mostradas en la Figura\(\PageIndex{11}\) y Figura\(\PageIndex{12}\) se pueden obtener las siguientes conclusiones:
- Dos picos de reducción y el inicio es de aproximadamente -0.75 eV (es decir, Figura\(\PageIndex{9}\) b).
- Un pico de oxidación con inicio de aproximadamente 0.85 eV (es decir, Figura\(\PageIndex{9}\) a).
- La brecha de banda calculada = 1.60 eV
En conclusión, hay muchas aplicaciones para el sistema CV, método eficiente, y la aplicación en el campo de la célula solar proporciona la información de banda gap para la investigación.
Aplicaciones de la voltametría cíclica en celdas de combustible de membrana de intercambio protónico
Introducción
Las celdas de combustible de membrana de intercambio de protones (PEMFC) son una alternativa prometedora a los motores de combustión tradicionales. Este método aprovecha la reacción exotérmica de oxidación de hidrógeno para generar energía y agua (Tabla\(\PageIndex{1}\)).
| Electrolito Acido | Potencial redox ácido en STP (V) | Electrolito básico | Potencial redox básico en STP (V) | |
| Semi-reacción del ánodo | \( 2H_{2} \rightarrow \ 4H^{+}\ +\ 4e^{-} \) | \( 2H_{2}\ +\ 4OH^{-} \rightarrow \ 4H_{2}O\ +\ 4e^{-} \) | ||
| Semi-reacción catódica | \( O_{2} + 4e^{-}\ +\ 4H^{+} \rightarrow \ 2H_{2}O \) | 1.23 | \( O_{2}\ +\ 4e^{-}\ +\ 2H_{2}O \rightarrow \ 4OH^{-} \) | 0.401 |
El PEMFC básico consiste en un ánodo y un cátodo separados por una membrana de intercambio de protones (Figura\(\PageIndex{13}\)). Esta membrana es un componente clave de la pila de combustible porque para que las reacciones del par redox ocurran con éxito, los protones deben poder pasar del ánodo al cátodo. La membrana en un PEMFC suele estar compuesta por Nafion, que es un ácido sulfónico polifluorado, y exclusivamente permite el paso de protones. Como resultado, los electrones y protones viajan del ánodo al cátodo a través de un circuito externo y a través de la membrana de intercambio de protones, respectivamente, para completar el circuito y formar agua.
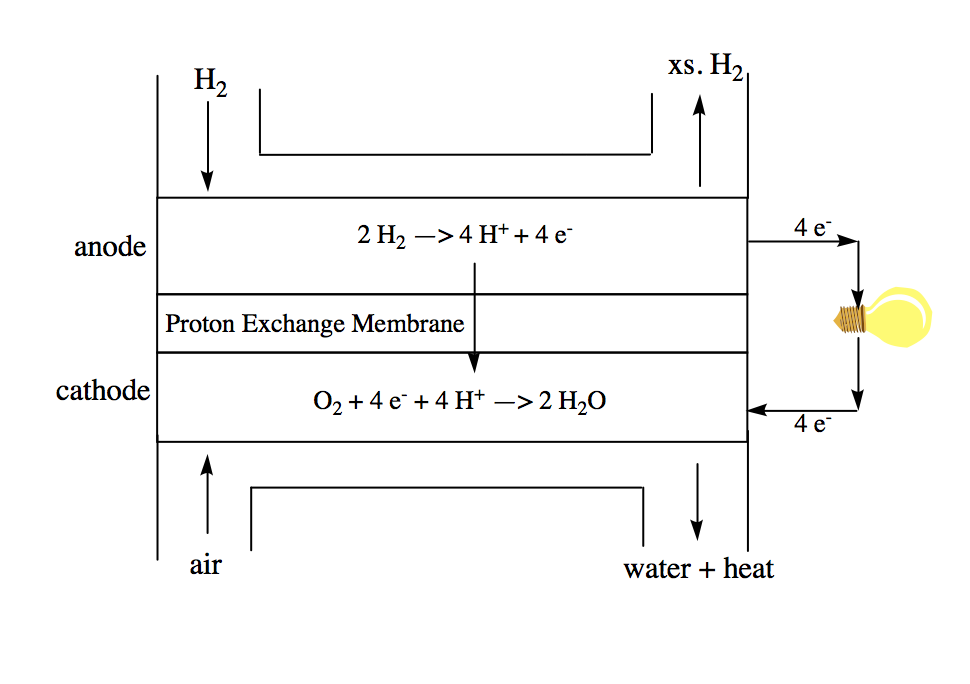
Los PEMFC presentan muchas ventajas en comparación con los motores de combustión tradicionales. Son más eficientes y tienen una mayor densidad energética que los combustibles fósiles tradicionales. Además, la pila de combustible en sí es muy simple con pocas o ninguna pieza móvil, lo que la hace duradera, confiable y muy silenciosa. Lo más importante, sin embargo, la operación de un PEMFC da como resultado cero emisiones ya que el único subproducto es el agua (Cuadro\(\PageIndex{2}\)). Sin embargo, el uso de PEMFC ha sido limitado debido a la lenta velocidad de reacción para la semi-reacción de reducción de oxígeno (ORR). Las velocidades de reacción, k°, para reacciones de reducción-oxidación como estas tienden a ser del orden de 10 -10 — 10 -9 donde 10 -10 es la velocidad de reacción más rápida y 10 -9 es la velocidad de reacción más lenta. En comparación con la semi-reacción de oxidación de hidrógeno (HOR), que tiene una velocidad de reacción de k° = 1x10 -10 cm/s, la velocidad de reacción para el ORR es k° ~ 1x10-9 cm/s, por lo que la ORR es la semi-reacción cinética limitante de la velocidad y su velocidad de reacción debe aumentarse para que los PEMFC sean viables alternativa a los motores de combustión. Debido a que la voltametría cíclica puede ser utilizada para examinar la cinética de la reacción ORR, es una técnica crítica en la evaluación de posibles soluciones a este problema.
| Ventajas | Desventajas |
| Más eficiente que la combustión | La media reacción de ORR es demasiado lenta para uso comercial |
| Mayor densidad energética que los combustibles fósiles | El combustible de hidrógeno no está fácilmente disponible |
| De larga duración | Debe manejarse la circulación del agua para mantener hidratada la membrana de intercambio de protones |
| Confiable | |
| Tranquilo | |
| Sin emisiones dañinas |
Voltamperometría cíclica
Visión general
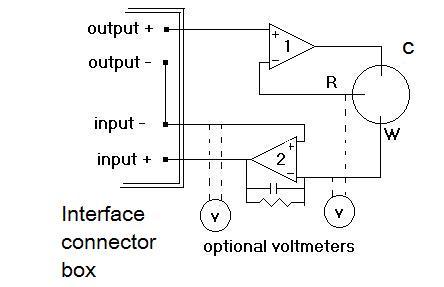
La voltametría cíclica es una técnica electroquímica clave que, entre sus otros usos, puede emplearse para examinar la cinética de las reacciones de oxidación-reducción en sistemas electroquímicos. Específicamente, los datos recopilados con voltametría cíclica pueden ser utilizados para determinar la velocidad de reacción. En su forma más simple, esta técnica requiere de una simple celda de tres electrodos y una figura de potenciostato\(\PageIndex{14}\).
Un potencial aplicado al electrodo de trabajo se varía linealmente con el tiempo y se mide la respuesta en la corriente Figura\(\PageIndex{14}\). Por lo general, el potencial es ciclado entre dos valores una vez en la dirección hacia adelante y una vez en la dirección inversa. Por ejemplo, en la Figura\(\PageIndex{15}\), el potencial es ciclado entre 0.8V y -0.2V con la exploración hacia adelante pasando de potencial positivo a negativo y la exploración inversa pasando de potencial negativo a positivo. Se pueden ajustar varios parámetros, incluyendo la velocidad de exploración, el número de ciclos de exploración y la dirección de la exploración potencial, es decir, si la exploración hacia adelante se mueve de voltajes positivos a negativos o viceversa. Para su publicación, los datos generalmente se recopilan a una velocidad de escaneo de 20 mV/s con al menos 3 ciclos de escaneo.
Lectura de un voltamograma
A partir de un experimento de voltametría cíclica, se obtendrá una gráfica llamada voltamograma. Debido a que tanto las semi-reacciones de oxidación como de reducción ocurren en la superficie del electrodo de trabajo, se observarán cambios pronunciados en la corriente cuando se produzca alguna de estas semirreacciones.Un voltamograma típico contará con dos picos donde un pico corresponde a la semirreacción de oxidación y el otro al media reacción de reducción. En una semi-reacción de oxidación en una celda electroquímica, los electrones fluyen desde las especies en solución al electrodo dando como resultado una corriente anódica, i a. Frecuentemente, este pico de oxidación aparece al escanear de potenciales negativos a positivos (Figura\(\PageIndex{16}\)). En una media reacción de reducción en una celda electroquímica, los electrones fluyen desde el electrodo a la especie en solución, dando como resultado una corriente catódica, i c. Este tipo de corriente se observa con mayor frecuencia al escanear de potenciales positivos a negativos. Cuando el reactivo de partida está completamente oxidado o completamente reducido, se alcanzan la corriente anódica pico, i pa, y la corriente catódica pico, i pc, respectivamente. Entonces, la corriente decae a medida que las especies oxidadas o reducidas salen de la superficie del electrodo. La forma de estos picos anódicos y catódicos se puede modelar con la ecuación de Nernst,\ ref {2}, donde número de electrones transferidos y E˚ '(potencial de reducción formal) = (E pa + E pc) /2
\[ E_{eq}\ =\ E^{\circ '} \ +\ (0.059/n)\ log\ ( [ reactant ] / [ product ] ) \label{2} \]
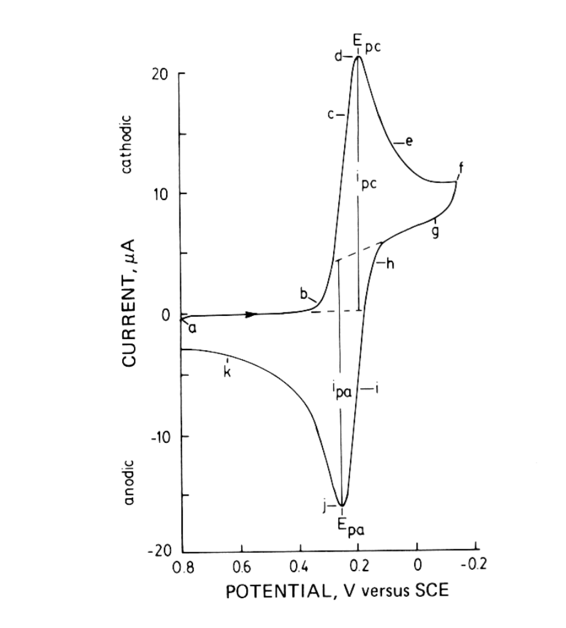
Valores Importantes del Voltamograma
Se pueden obtener varias piezas clave de información mediante el examen del voltamograma, incluyendo i pa, i pc, y los potenciales pico anódico y catódico. i pa e i pc sirven como medidas importantes de la actividad catalítica: cuanto mayores son las corrientes pico, mayor la actividad del catalizador. Los valores para i pa e i pc se pueden obtener a través de uno de dos métodos: examen físico de la gráfica o la ecuación de Randles-Sevick. Para determinar los potenciales de pico directamente a partir de la gráfica, se cruza una línea tangente vertical de la corriente pico con una línea base extrapolada. En contraste, la ecuación de Randles-Sevick utiliza información sobre el electrodo y los parámetros experimentales para calcular la corriente pico,\ ref {3}, donde A = área del electrodo; D = coeficiente de difusión; C = concentración; v = velocidad de barrido.
\[ i_{p} \ =\ (2.69x10^{5})n^{3/2}AD^{1/2}C\nu ^{12} \label{3} \]
El potencial pico anódico, E pa, y el potencial pico catódico, E pc, también se pueden obtener a partir del voltamograma determinando el potencial en el que ocurren i pa e i pc respectivamente. Estos valores son un indicador de la magnitud relativa de la velocidad de reacción. Si el intercambio de electrones entre los agentes oxidantes y reductores es rápido, forman un par electroquímicamente reversible. Estas parejas redox cumplen la relación: ΔE p = E pa — E pc ≡ 0.059/n. En contraste, una pareja no reversible tendrá un intercambio lento de electrones y ΔE p > 0.059/n Sin embargo, es importante señalar que ΔE p depende de la velocidad de barrido.
Análisis de Cinética de Reacción
Las ecuaciones de Tafel y Butler-Volmer permiten calcular la velocidad de reacción a partir de los datos de corriente-potencial generados por el voltamograma. En estos análisis, la velocidad de reacción se puede expresar como dos valores: k° e i o. k˚, la constante de velocidad estándar, es una medida de la rapidez con la que el sistema alcanza el equilibrio: cuanto mayor es el valor de k°, más rápida es la reacción. La densidad de corriente de intercambio, (i o) es el flujo de corriente en la superficie del electrodo en equilibrio: cuanto mayor es el valor de i o, más rápida es la reacción. Si bien se pueden usar tanto i o como k°, i o se usa con mayor frecuencia porque está directamente relacionado con el sobrepotencial a través de las ecuaciones de sobrepotencial actual y Butler-Volmer. Cuando la reacción está en equilibrio, k o e i o se relacionan por\ ref {4}, donde C o, eq y C R, eq = concentraciones de equilibrio de las especies oxidadas y reducidas respectivamente y a = factor de simetría.
\[ i_{O} \ =\ nFk^{\circ }C_{O, eq} ^{1-a} C_{R, eq} ^{a} \label{4} \]
Ecuación de Tafel
En su forma más simple, la ecuación de Tafel se expresa como\ ref {4}, donde a y b pueden ser una variedad de constantes. Cualquier ecuación que tenga la forma de\ ref {5} se considera una ecuación de Tafel.
\[ E-E^{\circ} \ =\ a\ +\ b\ log(i) \label{5} \]
Por ejemplo, la relación entre corriente, potencial, concentración de reactivos y productos, y k˚ puede expresarse como\ ref {6}, donde C O (0, t) y C R (0, t) = concentraciones de las especies oxidadas y reducidas respectivamente en un tiempo de reacción específico, F = constante de Faraday, R = constante de gas, y T = temperatura.
\[ C_{O}(0,t)\ -\ C_{R}(0,t)e^{ {[nf/RT] (E-E^{\circ } ) } } \ =\ [i/nFk^{\circ } ][e^{ {[anF/RT](E-E^{\circ } ) } } ] \label{6} \]
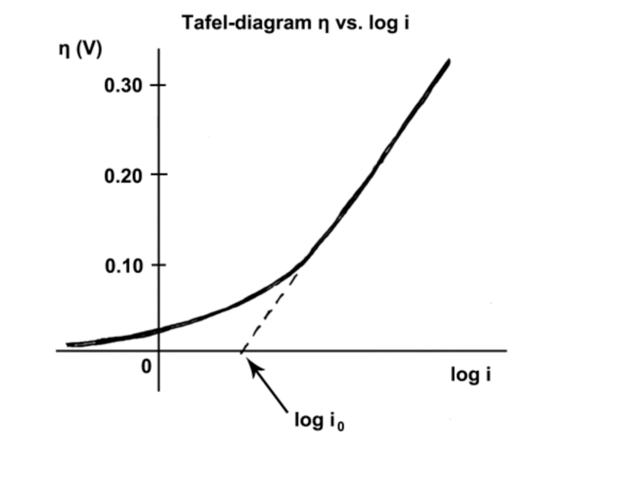
A sobrepotenciales muy grandes, esta ecuación se reduce a una ecuación de Tafel,\ ref {7}, donde a = - [RT/ (1-a) nF] ln (i o) y b = [RT/ (1-a) nF].
\[ E-E^{\circ } \ =\ [RT/(1-a)nF] ln(i)\ -\ [RT/(1-a)nF]ln(i_{0}) \label{7} \]
La relación lineal entre E-E˚ y log (i) puede explotarse para determinar i o mediante la formación de una parcela de Tafel (Figura\(\PageIndex{17}\)), E-E˚ versus log (i) .Las ramas anódica y catódica resultantes de la gráfica tienen pendientes de [(1-a) NF/2.3RT] y [-ANF/2.3RT], respectivamente. Una extrapolación de estas dos ramas da como resultado una intercepción y = log (i o). Así, esta gráfica relaciona directamente los datos potenciales y actuales recopilados por voltamperometría cíclica con i o.
Ecuación de Butler-Volmer
Si bien la ecuación de Butler-Volmer se asemeja a la ecuación de Tafel, y en algunos casos incluso puede reducirse a la formulación de Tafel, proporciona de manera única una relación directa entre i o y η. Sin simplificación, la ecuación de Butler-Volmer se conoce como el sobrepotencial actual\ ref {8}.
\[ i/i_{O}\ =\ C_{O}(0,t)/C_{O,eq}]e^{ { [anF/RT] (E-E^{\circ } ) } } \ -\ [ C_{R}(0,t)/C_{R,eq} ] e^{ { [ (1-a)nF/RT] (E-E^{\circ } ) } } \label{8} \]
Si la solución está bien agitada, se puede suponer que las concentraciones volumétricas y superficiales son iguales y\ ref {8} se puede reducir a la ecuación de Butler-Volmer,\ ref {9}.
\[ I\ =\ i_{O}[ e^{\{ [ anF/RT] (E-E^{\circ } )\} } - e^{ [ (1-a)nF/RT] (E-E^{\circ }) } ] \label{9} \]
Voltamperometría cíclica en la investigación de catálisis ORR
Catálisis de Platino
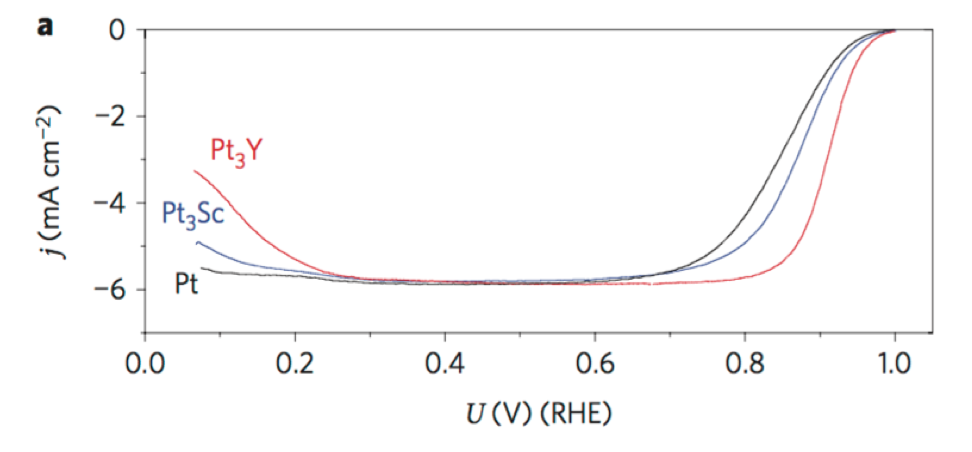
Si bien el tema de una velocidad de reacción de ORR lenta se ha abordado de muchas maneras, la mayoría de las veces se supera con el uso de catalizadores. Tradicionalmente, los catalizadores de platino han demostrado el mejor desempeño a 30 °C, el ORR i o sobre un catalizador de Pt es 2.8 x 10 -7 A/cm 2 comparado con el caso limitante de ORR donde i o = 1 x 10 -10 A/cm 2. El Pt es particularmente efectivo como catalizador para el ORR en PEMFC debido a que su energía de unión tanto para O como para OH es la más cercana al ideal de todos los metales a granel, su actividad es la más alta de todos los metales a granel, su selectividad para la adsorción de O 2 es cercana al 100%, y su estabilidad extrema bajo variedad de condiciones ácidas y básicas así como altos voltajes de operación Figura\(\PageIndex{18}\).
Catálisis compuesta Metal-Nitrogra-Carbono
Los catalizadores de metales no preciosos (NPMC) muestran un gran potencial para reducir el costo del catalizador sin sacrificar la actividad catalítica. Los mejores NPMC actualmente en desarrollo tienen una actividad y estabilidad de ORR comparables o incluso mejores que los catalizadores a base de platino en electrolitos alcalinos; en electrolitos ácidos, sin embargo, los NPMC funcionan significativamente peor que los catalizadores basados en platino.
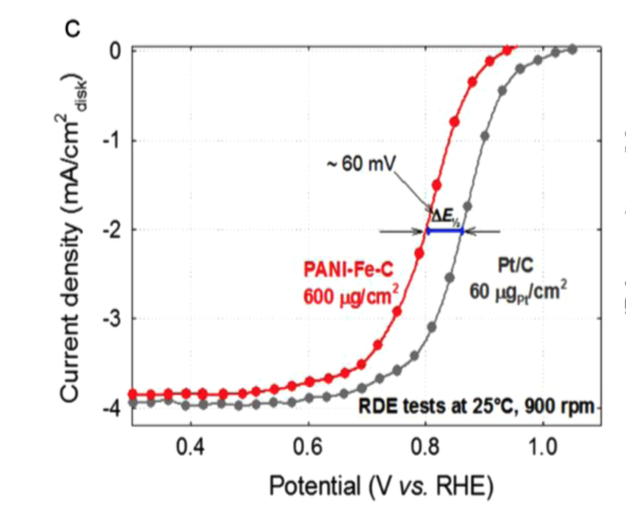
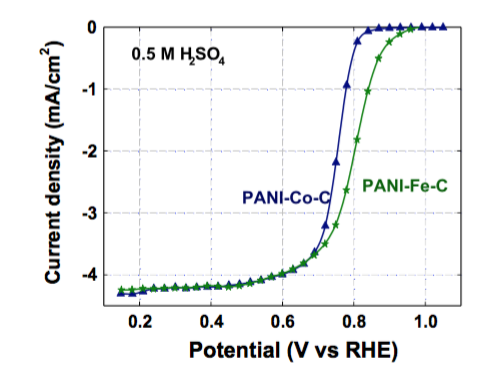
En particular, los catalizadores compuestos de metal de transición-nitrógeno-carbono (M-N-C) son el tipo de NPMC más prometedor. Los miembros de mayor rendimiento de este grupo catalizan el ORR a potenciales dentro de 60 mV de los catalizadores de platino de mayor rendimiento (Figura\(\PageIndex{19}\)). Adicionalmente, estos catalizadores tienen una excelente estabilidad: después de 700 horas a 0.4 V, no muestran ninguna degradación del desempeño. En una comparación de Pani-CO-C y PanI-Fe-C de alto rendimiento (PANI = polianilina), Zelenay y sus compañeros de trabajo utilizaron voltametría cíclica para comparar la actividad y el rendimiento de estos dos catalizadores en H 2 SO 4. Se encontró que el catalizador Co-Pani-C no tenía características de reducción-oxidación en su voltamograma, mientras que Fe-PANi-C se encontró que tenía dos picos redox a ~0.64 (Figura\(\PageIndex{20}\)). Estos picos de Fe-PANi-C tienen un ancho completo a la mitad máxima de ~100 mV, lo que es indicativo de la reducción-oxidación reversible de un electrón Fe 3+ /Fe 2+ (FWHM teórico = 96 mV). Zelenay y sus compañeros de trabajo también determinaron la densidad de corriente de intercambio usando el análisis de Tafel y encontraron que Fe-PANi-C tiene un i o (i o = 4 x 10 -8 A/cm 2) significativamente mayor en comparación con CopANi-C (i o = 5 x 10 -10 A/cm 2) en comparación con Copani-C (i o = ). Estas diferencias no solo demuestran la mayor actividad ORR de Fe-PANi-C cuando se compara con la Co-Pani-C, sino que también sugieren que los sitios activos ORR y los mecanismos de reacción son diferentes para estos dos catalizadores. Si bien se ha examinado la estructura de Fe-PANi-C (Figura\(\PageIndex{21}\)), la estructura de Co-Pani-C aún se está investigando.
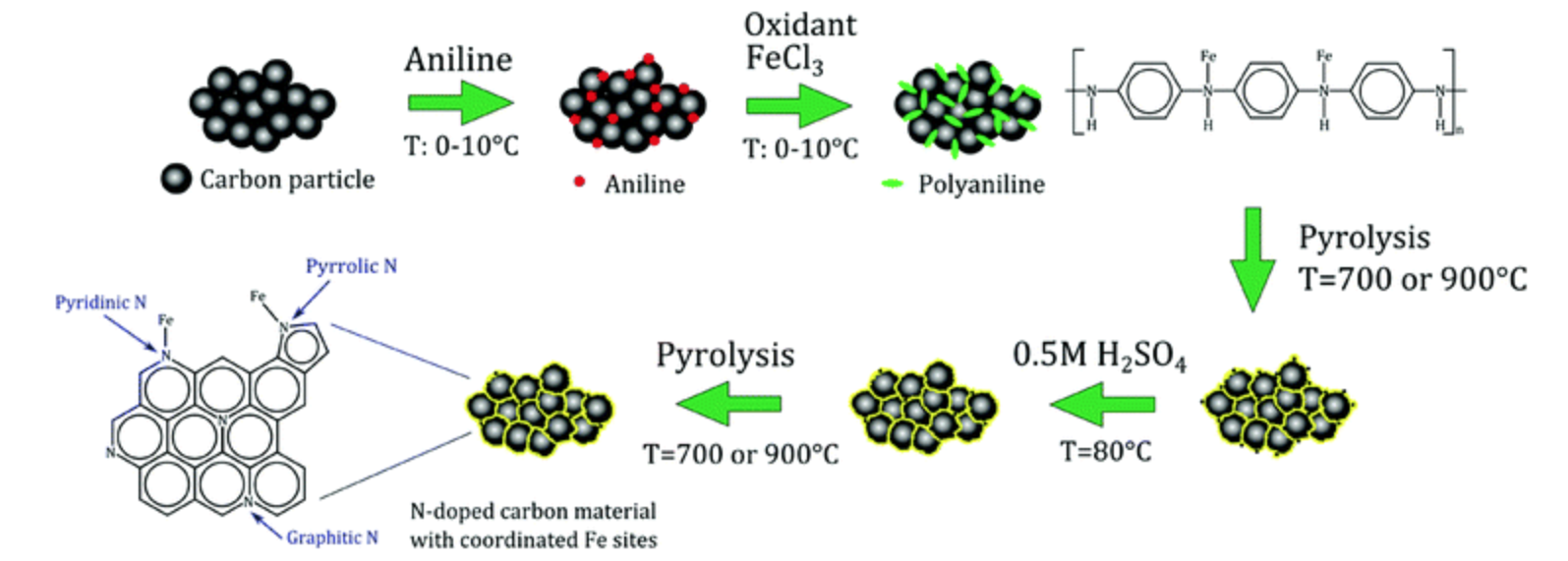
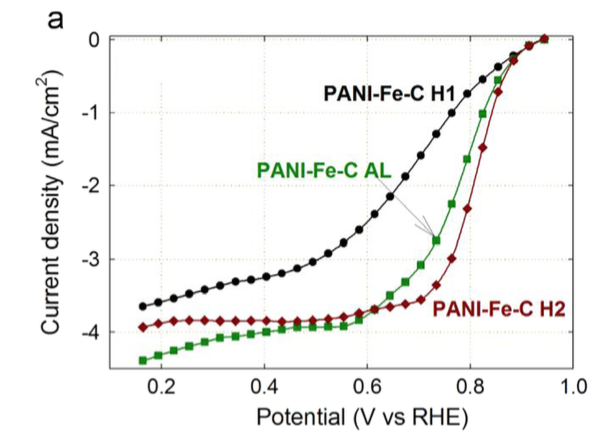
Mientras que la mayoría de los catalizadores M-N-C muestran alguna actividad ORR, la magnitud de esta actividad depende en gran medida de una variedad de factores; la voltametría cíclica es crítica en el examen de las relaciones entre cada factor y la actividad catalítica. Por ejemplo, la actividad de M-N-Cs es altamente dependiente del procedimiento sintético. En su examen en profundidad de los catalizadores de Fe-PANi-C, Zelenay y sus colaboradores optimizaron el procedimiento sintético para este catalizador examinando tres etapas de síntesis: el primer tratamiento de calentamiento, el paso de lixiviación con ácido y el segundo tratamiento de calentamiento. Su procedimiento sintético implicó la formación de una suspensión PANI-Fe-negro de carbón que se secó al vacío sobre un soporte de carbón. Luego, el catalizador intacto se sometió a un tratamiento de calentamiento de una hora seguido de lixiviación ácida y un tratamiento de calentamiento de tres horas. Los tratamientos de calentamiento se realizaron a 900˚C, que previamente se determinó que era la temperatura óptima para lograr la máxima actividad ORR (Figura\(\PageIndex{21}\)).
Para determinar los efectos de las etapas sintéticas sobre el catalizador intacto, los catalizadores Fe-PANi-C se analizaron por voltametría cíclica después del primer tratamiento térmico (HT1), después de la lixiviación ácida (AL) y después del segundo tratamiento térmico (HT2). En comparación con HT1, tanto los pasos AL como HT2 mostraron incrementos en la actividad catalítica. Adicionalmente, se encontró que HT2 aumentaba la actividad catalítica incluso más que la AL (Figura\(\PageIndex{22}\)). Con base en estos datos, Zelenay y sus compañeros de trabajo concluyeron que HT1 probablemente crea sitios activos en la superficie catalítica mientras que tanto la etapa de AL elimina impurezas, que bloquean los poros superficiales, para exponer sitios más activos. Sin embargo, también se sabe que esta etapa oxida parte del área catalítica. Por lo tanto, el aumento adicional de la actividad después de HT2 es probablemente el resultado de “reparar” la oxidación catalítica de la superficie.
Conclusión
Con nuevos avances en la investigación catalítica, los PEMFC se convertirán en una tecnología viable y ventajosa para el reemplazo de motores de combustión. El análisis de la actividad catalítica y la velocidad de reacción que proporciona la voltametría cíclica es crítico para comparar nuevos catalizadores con el catalizador actual de mayor rendimiento: Pt.
Cronocoulometría: una técnica para galvanoplastia
Fundamentos de Electroquímica
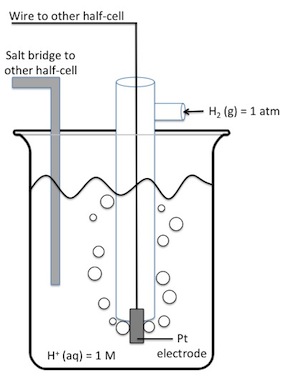
Una reacción química que implica un cambio en la carga de una especie química se denomina reacción electroquímica. Como su nombre indica, estas reacciones implican transferencia de electrones entre sustancias químicas. Muchas de estas reacciones ocurren espontáneamente cuando los diversos químicos entran en contacto entre sí. Para forzar que ocurra una reacción electroquímica no espontánea, se necesita proporcionar una fuerza impulsora. Esto se debe a que cada especie química tiene un potencial de reducción relativo. Estos valores proporcionan información sobre la capacidad del químico para tomar electrones adicionales. Por el contrario, podemos pensar si los potenciales de oxidación relativos, que indican la capacidad de un químico para regalar electrones. Es importante señalar que estos valores son relativos y necesitan definirse frente a una reacción de referencia. Una lista de potenciales de reducción estándar (estándar que indica medición contra el electrodo de hidrógeno normal como se ve en (Figura\(\PageIndex{23}\)) para medias reacciones electroquímicas comunes se da en la Tabla\(\PageIndex{3}\). Los sistemas electroquímicos no espontáneos, a menudo llamados celdas electrolíticas, como se mencionó anteriormente, requieren de una fuerza impulsora para que ocurra. Esta fuerza impulsora es un voltaje aplicado, que obliga a la reducción del químico que es menos probable que gane un electrón.
| Oxidante | Reductante | E° (V vs NHE) |
| 2H 2 O +2e - | H 2 (g) +2OH - | -0.8227 |
| Cu 2 O (s) H 2 O + 2e - | 2Cu (s) + 2OH - | -0.360 |
| Sn 4+ + 2e - | Sn 2+ | +0.15 |
| Cu 2+ + 2e - | Cu (s) | +0.337 |
| O 2 (g) + 2H + + 2e - | H 2 O 2 (aq) | +0.70 |
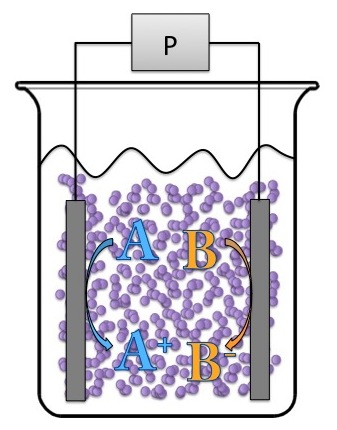
Diseño de una Célula Electroquímica
Un esquema de una celda electroquímica se ve en la Figura\(\PageIndex{24}\). Cualquier celda electroquímica debe tener dos electrodos: un cátodo, donde se produce la media reacción de reducción, y un ánodo, donde se produce la semirreacción de oxidación. Ejemplos de reacciones medias se pueden ver en la Tabla\(\PageIndex{3}\). Los dos electrodos están conectados eléctricamente de dos maneras: la solución electrolítica y el cable externo. La solución electrolítica típicamente incluye una pequeña cantidad del analito electroactivo (la especie química que realmente participará en la transferencia de electrones) y una gran cantidad de electrolito de soporte (las especies químicas que ayudan en el movimiento de la carga, pero que en realidad no están involucradas en los electrones transferencia. El cable externo proporciona una trayectoria para que los electrones viajen desde la semireacción de oxidación hasta la semirreacción de reducción. Como se mencionó anteriormente, cuando se está forzando a que ocurra una reacción electrolítica (no espontánea), se necesita aplicar un voltaje. Esto requiere que los cables estén conectados a un potenciostato. Como su nombre indica, un potenciostato controla el voltaje (es decir, “potentio” = potencial medido en voltios). Los componentes de una celda electroquímica y sus funciones también se dan en la Tabla\(\PageIndex{4}\).
| Componente | Función |
| Electrodo | Interfaz entre iones y electrones |
| Ánodo | Electrodo en el que tiene lugar la semireacción de oxidación |
| Cátodo | Electrodo en el que tiene lugar la media reacción de reducción |
| Solución electrolítica | Solución que contiene electrolito de soporte y analito electroactivo |
| Electrolito de soporte | No es parte del proceso faradaico; solo una parte del proceso capacitivo |
| Análito electroactivo | La especie química responsable de toda la corriente faradaica |
| Potenciostato | Fuente de voltaje CC; establece la diferencia de potencial entre el cátodo y el ánodo |
| Alambre | Conecta los electrodos al potenciostato |
Cronocoulometría: una técnica electroanalítica
Teoría
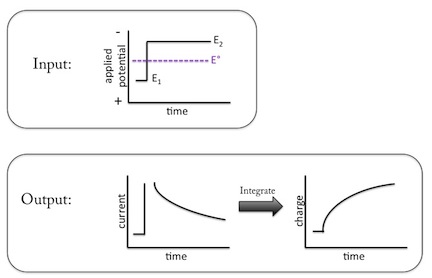
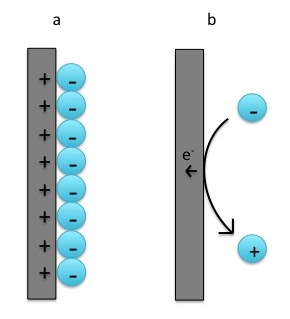
La cronocoulometría, como indica el nombre, es una técnica en la que se mide la carga (es decir, “coulometría”) en función del tiempo (es decir, “crono”). Existen diversos tipos de culombimetría. El que aquí se discute es la cululometría potenciostática en la que se establece el potencial (o voltaje) y, como resultado, la carga fluye a través de la celda. Los gráficos de ejemplo de entrada y salida se pueden ver en la Figura\(\PageIndex{25}\). La entrada es un paso potencial que abarca el potencial de reducción de las especies electroactivas. Si esta etapa de potencial se realiza en una celda electroquímica que no contiene especies electroactivas, solo fluirá corriente capacitiva (Figura\(\PageIndex{26}\)), en la que los iones migran de tal manera que las cargas se alinean (positivas junto a negativas, pero no se transfiere carga alguna. Sin embargo, una vez que se introduce una especie electroactiva en el sistema, la corriente faradaica comienza a fluir. Esta corriente es el resultado de la transferencia de electrones entre el electrodo y las especies electroactivas.
Galvanoplastia: una aplicación de cronocoulometría
La galvanoplastia es un proceso electroquímico que utiliza técnicas como la cronocoulometría para electrodepositar un químico cargado de una solución como un químico neutro en la superficie de otro químico. Estos productos químicos suelen ser metales. La ciencia de la galvanoplastia se remonta a principios del siglo XIX cuando Luigi Valentino Brugnatelli (Figura\(\PageIndex{27}\)) galvanizó oro de solución a metales de plata. A mediados del siglo XIX, el proceso de galvanoplastia fue patentado por los primos George y Henry Elkington (Figura\(\PageIndex{28}\)). Los Elkingtons trajeron bienes galvanizados a las masas al producir productos de consumo como joyas artificiales y otros artículos conmemorativos (Figura\(\PageIndex{29}\)).



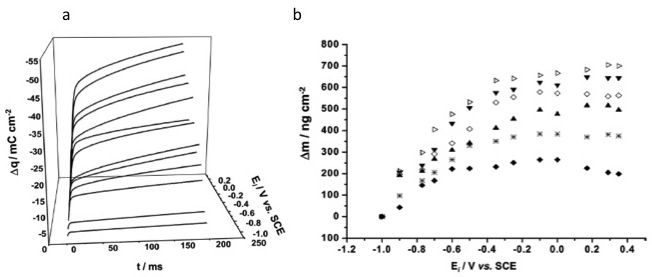
Estudios científicos recientes han tomado interés en estudiar la galvanoplastia. Trejo y compañeros de trabajo han demostrado que se puede utilizar una microbalanza de cuarzo para medir el cambio de masa a lo largo del tiempo durante la electrodeposición mediante cronocoulometría. La figura\(\PageIndex{30}\) a muestra la carga transferida en varias etapas potenciales. La Figura\(\PageIndex{30}\) b muestra el cambio de masa en función del escalón potencial. Es claro que la magnitud de la etapa potencial está directamente relacionada con la cantidad de carga transferida y consecuentemente con la masa de las especies electroactivas depositadas.
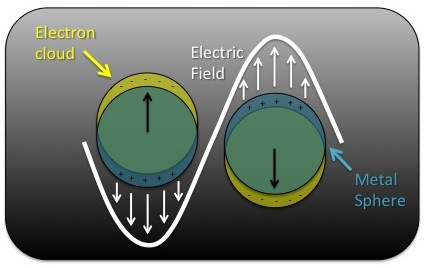
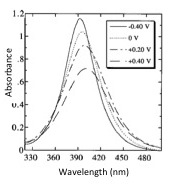
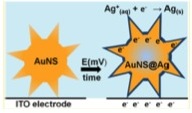
Se ha estudiado el efecto de la galvanoplastia mediante cronocoulometría sobre la resonancia de plasmón superficial localizada (LSPR) sobre nanopartículas metálicas. Un LSPR es la oscilación colectiva de electrones inducida por un campo eléctrico (Figura\(\PageIndex{31}\)). En diversos estudios realizados por Mulvaney y compañeros de trabajo, se observó un claro efecto sobre la frecuencia de LSPR a medida que se aplicaron potenciales (Figura\(\PageIndex{32}\)). En estudios iniciales, no se reportó evidencia de galvanoplastia. En estudios más recientes del mismo grupo, se demostró que las nanopartículas podrían ser electrochapadas mediante cronocoulometría (Figura\(\PageIndex{33}\). Tales desarrollos pueden conducir a una expansión de las aplicaciones tanto de galvanoplastia como de plasmónica.