3.6: Electroforesis Capilar
- Page ID
- 71206

La electroforesis capilar (CE) abarca una familia de técnicas de separación electrocinética que utiliza un campo eléctrico aplicado para separar analitos en función de su carga y tamaño. El principio básico se basa en el de la electroforesis, que es el movimiento de las partículas en relación con un fluido (electrolito) bajo la influencia de un campo eléctrico. El padre fundador de la electroforesis, Arne W. K. Tiselius (Figura\(\PageIndex{1} a \)), utilizó por primera vez la electroforesis para separar proteínas, y luego ganó el Premio Nobel de Química en 1948 por su trabajo tanto en electroforesis como en análisis de adsorción. Sin embargo, fue Stellan Hjerten (Figura\(\PageIndex{1} b\)) quien trabajó bajo Arne W. K. Tiselius, quien fue pionero en el trabajo en CE en 1967, aunque CE no fue bien reconocido hasta 1980 cuando James W. Jorgenson (Figura\(\PageIndex{1} c \)) y Krynn D. Lukacs publicaron una serie de artículos describiendo esta nueva técnica.

Descripción general del instrumento
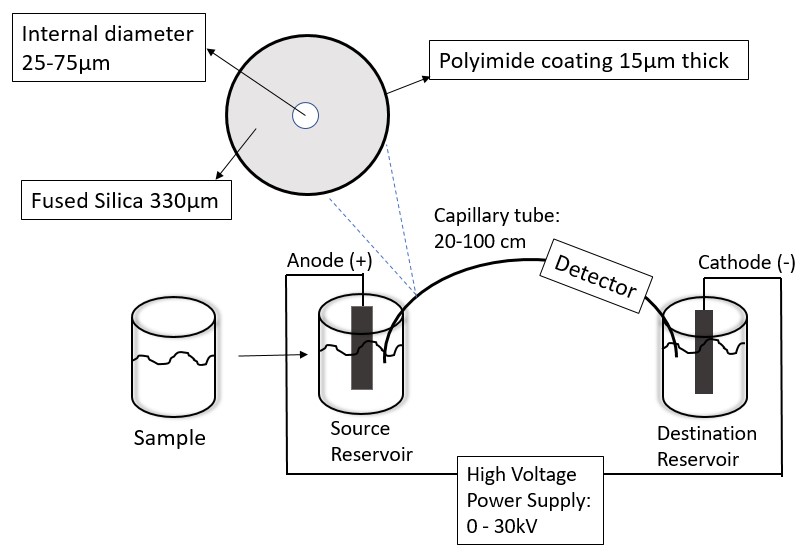
Los componentes principales de la CE se muestran en la Figura\(\PageIndex{2}\). El circuito eléctrico del CE es el corazón del instrumento.
Métodos de inyección
Las muestras que se estudian en CE son principalmente muestras líquidas. Una columna capilar típica tiene un diámetro interno de 50 μm y una longitud de 25 cm. Debido a que la columna solo puede contener una cantidad mínima de tampón de ejecución, solo se pueden probar pequeños volúmenes de muestra (nL a μL). Las muestras se introducen principalmente por dos métodos de inyección: inyección hidrodinámica y electrocinética. Los dos métodos se muestran en la Tabla\(\PageIndex{1}\) Una desventaja de la inyección electrocinética es que la composición de la muestra inyectada puede no ser la misma que la composición de la muestra original. Esto se debe a que el método de inyección depende de la movilidad electroforética y electroosmótica de las especies en la muestra. Sin embargo, ambos métodos de inyección dependen de la temperatura y la viscosidad de la solución. Por lo tanto, es importante controlar ambos parámetros cuando se desea un volumen reproducible de inyecciones de muestra. Es recomendable utilizar estándares internos en lugar de estándares externos al realizar análisis cuantitativos sobre las muestras ya que es difícil controlar tanto la temperatura como la viscosidad de la solución.
| Métodos de inyección | Principio de funcionamiento |
|---|---|
| Inyección hidrodinámica | El vial de muestra se encierra en una cámara con un extremo de columna capilar fija sumergido en ella. Luego se aplica presión a la cámara por un periodo fijo para que la muestra pueda ingresar al capilar. Después de que se haya introducido la muestra, se retira el capilar y luego se vuelve a sumergir en el depósito fuente y se lleva a cabo la separación. |
| Inyección electrocinética | La muestra se encierra en una cámara con un extremo de columna capilar sumergido en ella con un electrodo presente. Se aplica el campo eléctrico, y las muestras ingresan al capilar. Después de que se haya introducido la muestra, se retira el capilar y luego se vuelve a sumergir en el depósito fuente y se lleva a cabo la separación. |
Columna
Una vez inyectadas las muestras, se utiliza la columna capilar como medio principal para separar los componentes. La columna capilar utilizada en CE comparte las mismas características que la columna capilar utilizada en cromatografía de gases (GC); sin embargo, los componentes más críticos de la columna CE son:
- el diámetro interno del capilar,
- la longitud total del capilar,
- la longitud de la columna desde el inyector hasta el detector.
Tampón Solvente
El tampón disolvente transporta la muestra a través de la columna. Es crucial emplear un buen búfer ya que un experimento de CE exitoso depende de esto. CE se basa en la separación de cargas en un campo eléctrico. Por lo tanto, el tampón debe sostener la carga preexistente sobre el analito o permitir que el analito obtenga una carga, y es importante considerar el pH del tampón antes de usarlo.
Voltaje aplicado (kV)
El voltaje aplicado es importante en la separación de los analitos ya que impulsa el movimiento del analito. Es importante que no sea demasiado alto ya que puede convertirse en una preocupación de seguridad.
Detectores
Los analitos que se han separado después de aplicar el voltaje pueden ser detectados por muchos métodos de detección. El método más común es la absorbancia UV-Visible. La detección se realiza a través del capilar con una pequeña porción del capilar actuando como célula de detección. La celda de detección en tubo generalmente se hace ópticamente transparente raspando el recubrimiento de poliimida y recubriéndolo con otro material ópticamente transparente para que el capilar no se rompa fácilmente. Para las especies que no tienen cromóforo, se puede agregar un cromóforo a la solución tampón. Cuando el analito pasa, habría una disminución en la señal. Esta señal disminuida corresponderá a la cantidad de analito presente. Otras técnicas de detección comunes que se pueden emplear en CE son la fluorescencia y la espectrometría de masas (EM).
Teoría
En CE, la muestra se introduce en el capilar por los métodos mencionados anteriormente. Luego se aplica un alto voltaje provocando que los iones de la muestra migren hacia el electrodo en el reservorio de destino, en este caso, el cátodo. La migración y separación de los componentes de la muestra están determinadas por dos factores, la movilidad electroforética y la movilidad electroosmótica.
Movilidad Electroforética
La movilidad electroforética,\(μ_{ep}\), depende inherentemente de las propiedades del soluto y del medio en el que se mueve el soluto. Esencialmente, es un valor constante, que puede ser calculado como dado por\ ref {1} donde\(q\) está la carga del soluto,\(η\) es la viscosidad del tampón y\(r\) es el radio del soluto.
\[ \mu _{ep} = \dfrac{q}{6\pi \eta r} \label{1} \]
La velocidad electroforética,\(v_{ep}\), depende de la movilidad electroforética y del campo eléctrico aplicado,\(E\) (\ ref {2}).
\[ \nu _{ep} = \mu _{ep} E \label{2} \]
Así, cuando los solutos tienen una relación de carga a tamaño mayor, la movilidad electroforética y la velocidad aumentarán. Los cationes y el anión se moverían en direcciones opuestas correspondientes al signo de la movilidad electroforética con es resultado de su carga. Así, las especies neutras que no tienen carga no tienen movilidad electroforética.
Movilidad Electroosmótica
El segundo factor que controla la migración del soluto es el flujo electroosmótico. Con carga cero, se espera que las especies neutras permanezcan estacionarias. Sin embargo, en condiciones normales, la solución tampón también se mueve hacia el cátodo. La causa del flujo electroosmótico es la doble capa eléctrica que se desarrolla en la interfaz de la solución de sílice.
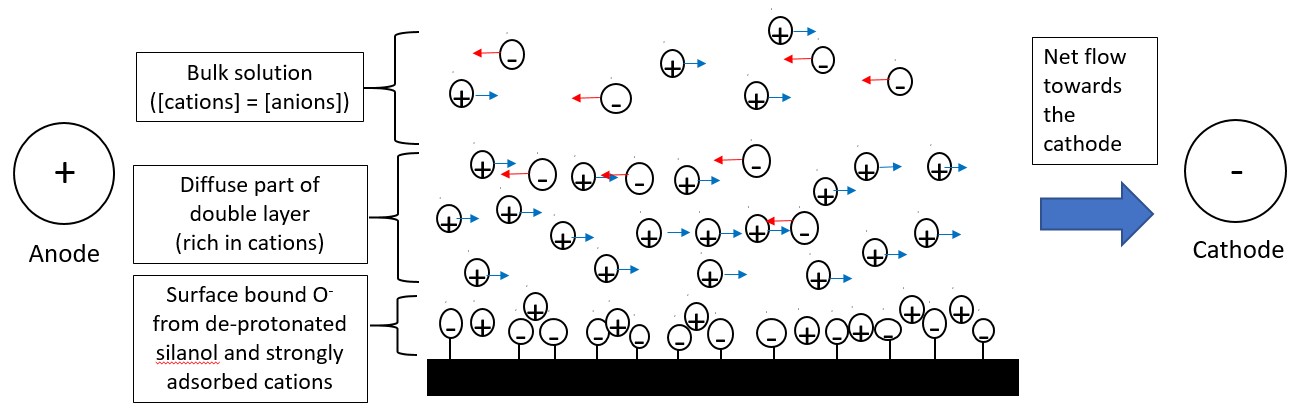
A pH superior a 3, los abundantes grupos silanol (-OH) presentes en la superficie interna del capilar de sílice, desprotonan para formar iones silanato cargados negativamente (-SiO -). Los cationes presentes en la solución tampón serán atraídos por los iones silanato y algunos de ellos se unirán fuertemente a ella formando una capa fija. La formación de la capa fija solo neutraliza parcialmente la carga negativa en las paredes capilares. De ahí que más cationes que aniones estarán presentes en la capa adyacente a la capa fija, formando la capa difusa. La combinación de la capa fija y la capa difusa se conoce como la doble capa como se muestra en la Figura\(\PageIndex{3}\). Los cationes presentes en la capa difusa migrarán hacia el cátodo, ya que estos cationes están solvatados la solución también fluirá con él, produciendo el flujo electroosmótico. Los aniones presentes en la capa difusa están solvatados y se moverán hacia el ánodo. Sin embargo, como hay más cationes que aniones, los cationes empujarán los aniones junto con él en la dirección del cátodo. De ahí que el flujo electroosmótico se mueva en la dirección del cátodo.
La movilidad electroosmótica, μ eof, se describe por\ ref {3} donde es el potencial zeta, ε es la constante dieléctrica del tampón y η es la viscosidad del tampón. La velocidad electroosmótica, v eof, es la velocidad a la que el búfer se mueve a través del capilar viene dada por\ ref {4}.
\[ \mu _{eof} \ =\ \frac{\zeta \varepsilon }{4\pi \eta } \label{3} \]
\[ \nu _{eof}\ =\ \mu _{eof}E \label{4} \]
Potencial Zeta
El potencial zeta, también conocido como potencial electrocinético es el potencial eléctrico en la interfaz de la doble capa. De ahí que en nuestro caso, es el potencial de la capa difusa el que se encuentra a una distancia finita de la pared capilar. El potencial zeta se ve afectado principalmente y es directamente proporcional a dos factores:
- El espesor de la doble capa. Una mayor concentración de cationes posiblemente debido a un aumento en la fuerza iónica del tampón conduciría a una disminución en el grosor de la doble capa. A medida que disminuye el grosor de la doble capa, el potencial zeta disminuiría lo que resulta en la disminución del flujo electroosmótico.
- La carga en las paredes capilares. Una mayor densidad de los iones silanato corresponde a un mayor potencial zeta. La formación de iones silanato depende del pH. De ahí que a pH menor a 2 hay una disminución en el potencial zeta y el flujo electroosmótico ya que el silanol existe en su forma protonada. Sin embargo, a medida que aumenta el pH, se forman más iones de silanato provocando un aumento en el potencial zeta y por lo tanto, el flujo electroosmótico.
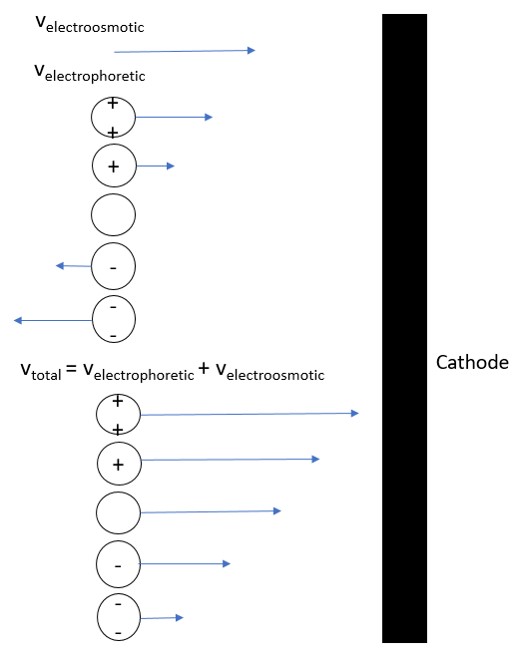
Orden de Elución
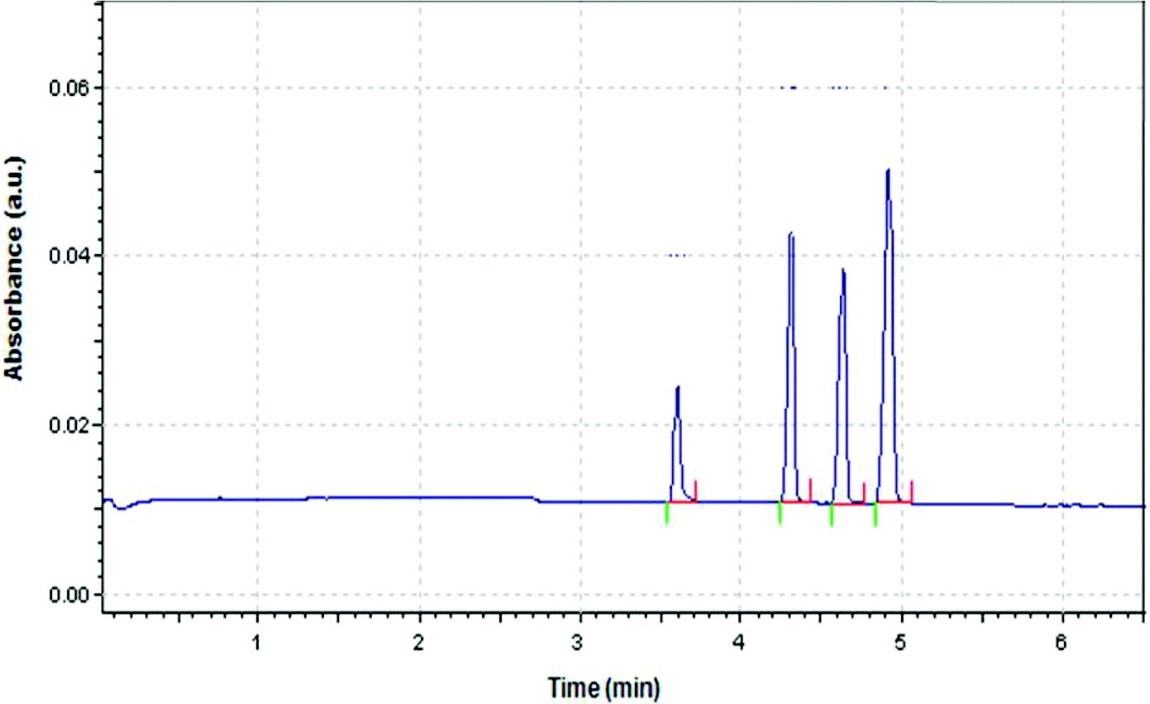
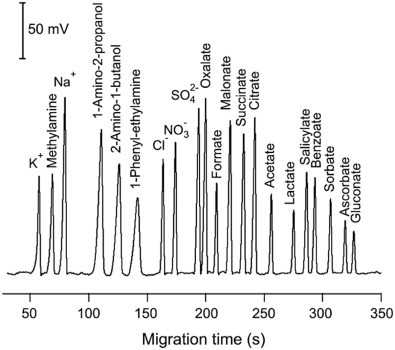
El flujo electroosmótico del tampón es generalmente mayor que el flujo electroforético de los analitos. De ahí que incluso los aniones se moverían al cátodo como se ilustra en la Figura\(\PageIndex{4}\). Los cationes pequeños y altamente cargados serían los primeros en eluir antes que los cationes más grandes con menor carga. Esto es seguido por la especie neutra que eluye como una banda en el medio. Los aniones más grandes con baja carga eluyen a continuación y, por último, el anión pequeño altamente cargado tendría el tiempo de elución más largo. Esto se retrata claramente en el electroferograma de la Figura\(\PageIndex{5}\).
Optimización del experimento CE
Existen varios componentes que se pueden variar para optimizar el electroferograma obtenido de CE. Por lo tanto, para cualquier configuración dada se deben conocer ciertos parámetros:
- la longitud total del capilar (L),
- la longitud que recorren los solutos desde el inicio hasta el detector (l),
- el voltaje aplicado (V).
Reducción en el tiempo de migración, t mn
Para acortar el tiempo de análisis, se puede usar un voltaje más alto o se puede usar un tubo capilar más corto. Sin embargo, es importante tener en cuenta que el voltaje no puede ser arbitrariamente alto ya que conducirá a un calentamiento por julios. Otra posibilidad es aumentar μ eof aumentando el pH o disminuyendo la fuerza iónica del tampón,\ ref {5}.
\[ t_{mn} \ =\ \frac{1\ L}{(\mu _{ep} \ +\ \mu_{eof}) V } \label{5} \]
Eficiencia
En cromatografía, la eficiencia viene dada por el número de placas teóricas, N. En CE, existe un parámetro similar,\ ref {6} donde D es el coeficiente de difusión del soluto. Incremento de eficiencia s con un incremento en el voltaje aplicado ya que el soluto pasa menos tiempo en el capilar habrá menos tiempo para que el soluto se difunda. Generalmente, para CE, N será muy grande.
\[ N\ =\frac{1^{2}}{2Dt_{mn}} = \frac{\mu _{tot} V l}{2DL} \label{6} \]
Resolución entre dos picos
La resolución entre dos picos, R, se define por\ ref {7} donde Δv es la diferencia de velocidad de dos solutos y es la velocidad promedio de dos solutos.
\[ R= \frac{\sqrt{N} }{4} \times \frac{\Delta v}{ \tilde{\nu } } \label{7} \]
Sustituyendo la ecuación por N da\ ref {8}
\[ R\ = 0.177(\mu _{ep,1} \ -\ \mu _{ep,2}) \sqrt{ \frac{V}{D(\nu _{av} + \mu _{eof})} } \label{8} \]
Por lo tanto, al aumentar el voltaje aplicado, V, se incrementará la resolución. Sin embargo, no es muy efectivo ya que un aumento de 4 veces en el voltaje aplicado solo daría un aumento de 2 veces en la resolución. Además, el incremento en N, el número de placas teóricas resultaría en una mejor resolución.
Selectividad
En cromatografía, la selectividad, α, se define como la relación de los dos factores de retención del soluto. Esto es lo mismo para CE,\ ref {9}, donde t 2 y t 1 son los tiempos de retención para los dos solutos tal que, α es más de 1.
\[ \alpha =\frac{t_{2}}{t_{1}} \label{9} \]
La selectividad se puede mejorar ajustando el pH de la solución tampón. El propósito es cambiar la carga de las especies que se eluyen.
Comparación entre CE y HPLC
La CE a diferencia de la cromatografía líquida de alto rendimiento (HPLC) acomoda muchas muestras y tiende a tener una mejor resolución y eficiencia. En la Tabla se da una comparación entre los dos métodos\(\PageIndex{2}\).
| CE | HPLC |
|---|---|
| Selección más amplia del analito a analizar | Limitado por la solubilidad de la muestra |
| Mayor eficiencia, sin término de transferencia de masa estacionaria ya que no hay fase estacionaria | La eficiencia se reduce debido al término de transferencia de masa estacionaria (equilibrio entre la fase estacionaria y móvil) |
| El perfil de flujo electroosmótico en el capilar es plano, por lo que no se ensancha la banda. Mejor resolución de picos y picos más nítidos | Perfil de flujo laminar redondeado que es común en sistemas impulsados por presión como HPLC. Resultando en picos más amplios y menor resolución |
| Se puede acoplar a la mayoría de los detectores dependiendo de la aplicación | Algunos detectores requieren que se cambie el disolvente y la modificación previa de la muestra antes del análisis |
| Mayor capacidad pico ya que utiliza un número muy grande de placas teóricas, N | La capacidad máxima se reduce ya que N no es tan grande |
| Se utilizan altos voltajes al realizar el experimento | No hay necesidad de alto voltaje |
Cromatografía Micelar Electrocinética
CE permite la separación de partículas cargadas, y se compara principalmente con la cromatografía iónica. Sin embargo, no se realiza ninguna separación para especies neutras en CE. Así, se puede utilizar una técnica CE modificada denominada cromatografía electrocinética micelar (MEKC) para separar neutrales en función de su tamaño y su afinidad con la micela. En MEKC, se agregan especies de surfactantes a la solución tampón a una concentración a la que se formarán micelas. Un ejemplo de un tensioactivo es el dodecilsulfato de sodio (SDS) como se ve en la Figura\(\PageIndex{6}\)
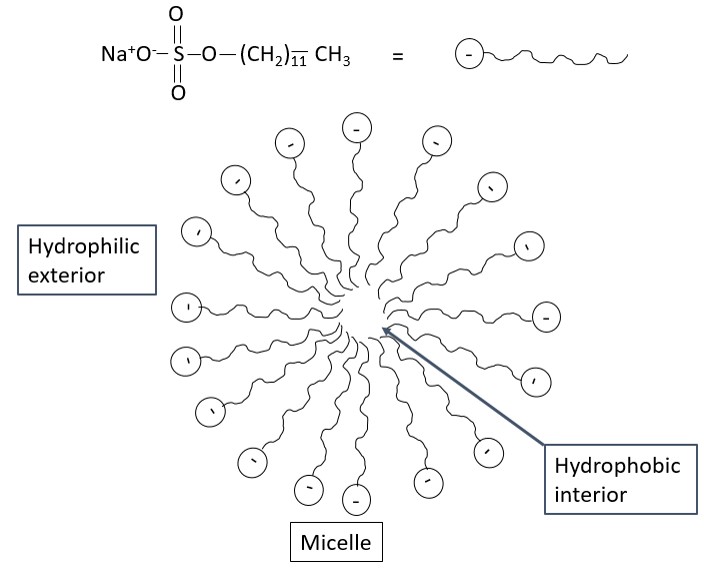
Las moléculas neutras están en equilibrio dinámico entre la solución a granel y el interior de la micela. En ausencia de la micela la especie neutra alcanzaría el detector a t 0 pero en presencia de la micela, alcanza el detector a t mc, donde t mc es mayor que t 0. Cuanto más tiempo permanezca la molécula neutra en la micela, mayor será el tiempo de migración. Así, especies neutras no polares pequeñas que favorecen la interacción con el interior de la micela tardarían más tiempo en llegar al detector que una especie polar grande. Se pueden agregar tensioactivos aniónicos, catiónicos e iónicos zwitter para cambiar el coeficiente de partición de las especies neutras. Los tensioactivos catiónicos resultarían en micelas positivas que se moverían en la dirección del flujo electroosmótico. Esto le permite moverse más rápido hacia el cátodo. Sin embargo, debido a la rápida migración, es posible que se dé un tiempo insuficiente para que las especies neutras interactúen con la micela dando como resultado una mala separación. Por lo tanto, se deben considerar todos los factores antes de elegir el surfactante adecuado para usar. El mecanismo de separación entre MEKC y cromatografía líquida es el mismo. Ambos dependen del coeficiente de partición de las especies entre la fase móvil y la fase estacionaria. La principal diferencia radica en la fase pseudo estacionaria en MEKC, las micelas. La micela que puede considerarse la fase estacionaria en MEKC se mueve a una velocidad más lenta que los iones móviles.
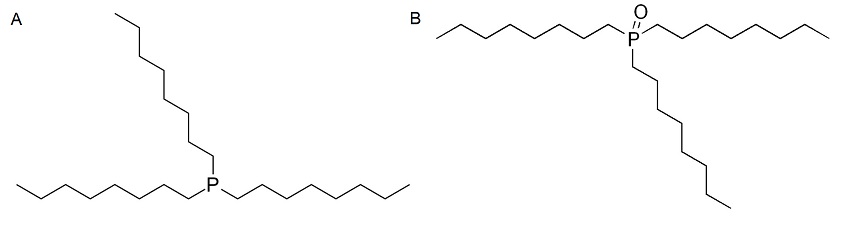
Los puntos cuánticos (QD) son nanocristales semiconductores que se encuentran en el rango de tamaño de 1-10 nm, y tienen diferente movilidad electroforética debido a sus diferentes tamaños y carga superficial. Se puede usar CE para separar y caracterizar tales especies, y se ha desarrollado un método para caracterizar y separar CdSe QD en el medio acuoso. Los QD se sintetizaron con una capa externa de trioctilfosfina (TOP, Figura\(\PageIndex{7} a\)) y óxido de trioctilfosfina (TOPO, Figura\(\PageIndex{7} b\)), haciendo que la superficie del QD sea hidrófoba. La solución electrolítica de fondo utilizada fue SDS, con el fin de hacer que los QDs sean solubles en agua y formar un complejo QD-TOPO/TOP-SDS. Se utilizaron diferentes tamaños de CdSe y la separación fue con respecto a la relación carga-masa de los complejos. Se concluyó del estudio que cuanto mayor era el núcleo de CdSe (es decir, cuanto mayor es la relación carga-masa) eluyó el último. El electroferograma del estudio se muestra en la Figura\(\PageIndex{8}\) a partir de la cual es visible que se había producido una buena separación mediante el uso de CE. Se utilizó detección de fluorescencia inducida por láser, el sistema tampón fue SDS y el pH del sistema configurado se fijó en 6.5. El pH es muy importante en este caso ya que la estabilidad del sistema y la separación depende de él.