
4.2: Espectroscopia IR
- Page ID
- 71070

Preparación de muestras IR: una guía práctica
La espectroscopia infrarroja se basa en vibraciones moleculares causadas por la oscilación de dipolos moleculares. Los enlaces tienen vibraciones características dependiendo de los átomos en el enlace, el número de enlaces y la orientación de esos enlaces con respecto al resto de la molécula. Así, diferentes moléculas tienen espectros específicos que pueden ser recolectados para su uso en distinguir productos o identificar una sustancia desconocida (hasta cierto punto).
La recolección de espectros a través de este método va alrededor de una de tres formas generales. Los mulls de Nujol y los pellets prensados se utilizan típicamente para recolectar espectros de sólidos, mientras que las celdas de película delgada se utilizan para la espectroscopia IR en fase de solución. También se pueden obtener espectros de gases pero no se discutirán en esta guía.
Materiales ópticos infrarrojos y manejo
Si bien está todo bien y maravilloso que las sustancias se puedan caracterizar de esta manera todavía hay que poder retener las sustancias dentro del instrumento y preparar adecuadamente las muestras. En un espectrómetro infrarrojo (Figura\(\PageIndex{1}\))
la muestra a analizar se mantiene frente a un rayo láser infrarrojo, para ello, la muestra debe estar contenida en algo, en consecuencia esto significa que el mismo recipiente en el que se encuentra la muestra absorberá parte del haz infrarrojo.

Esto se complica un tanto por el hecho de que todos los materiales tienen algún tipo de vibración asociada a ellos. Por lo tanto, si el portamuestras tiene una ventana óptica hecha de algo que absorbe cerca de donde lo hace su muestra, la muestra podría no ser distinguible de la ventana óptica del portamuestras. El rango que no está bloqueado por una fuerte absorbancia se conoce como ventana (no debe confundirse con los materiales ópticos de la celda).
Las ventanas son un factor importante a considerar a la hora de elegir el método para realizar un análisis, ya que se ve en la Tabla\(\PageIndex{1}\) hay una serie de materiales diferentes cada uno con sus propios espectros de absorción característicos y propiedades químicas. Tenga en cuenta estos factores al realizar análisis y se guardará una muestra preciosa. Para la mayoría de los compuestos orgánicos, el NaCl funciona bien aunque es susceptible al ataque de la humedad. Para los complejos de coordinación de metal, KBr o CSi normalmente funcionan bien debido a sus grandes ventanales. Si el dinero no es un problema entonces el diamante o el zafiro se pueden usar para las placas.
| Material | Rangos Transparentes (cm -1) | Solubilidad | Notas |
|---|---|---|---|
| NaCl | 40,000 - 625 | H 2O | Fácil de pulir, higroscópico |
| Vidrio de sílice | 55,000-3,000 | HF | Atacado por HF |
| Cuarzo | 40,000-2,500 | HF | Atacado por HF |
| Zafiro | 20,000-1,780 | - | Fuerte |
| Diamante | 40,000-2,500 y 1,800-200 | - | Muy fuerte, caro, duro, inútil para pellets |
| CaF 2 | 70,000-1,110 | Ácidos | Atacado por ácidos, evitar sales de amonio |
| BaF 2 | 65,000-700 | - | Evite las sales de amonio |
| ZnSe | 10,000 - 550 | Ácidos | Frágil, atacado por ácidos |
| AgCl | 25,000-400 | - | Suave, sensible a la luz. |
| KCl | 40,000-500 | H 2 O, Et 2 O, acetona | Higroscópico, suave, fácil de pulir, comúnmente utilizado en la fabricación de pellets. |
| KBr | 40,000-400 | H 2 O, EtOH | Higroscópico, suave, fácil de pulir, comúnmente utilizado en la fabricación de pellets. |
| CSBr | 10,000-250 | H 2 O, EtOH, acetona | Higroscópico suave |
| CSi | 10,000-200 | H 2 O, EtOH, MeOH, acetona | Higroscópico, suave. |
| Teflón | 5,000-1,200; 1,200-900 | - | Inerte, desechable |
| Polietileno | 4,000-3.000; 2,800-1,460; 1,380 - 730; 720- 30 | - | Inerte, desechable |
El manejo adecuado de estas placas asegurará que tengan una larga vida útil. Aquí sigue algunos consejos simples sobre cómo manejar las placas:
- Evite el contacto con disolventes en los que las placas sean solubles.
- Mantener las placas en un desecador, cuanto menos agua mejor, aunque las placas sean insolubles en agua.
- Asa con guantes, guantes limpios.
- Evite limpiar las placas para evitar que se rayen.
Dicho esto, estas sencillas pautas probablemente reducirán la mayoría de los daños que pueden ocurrir a una placa simplemente sujetándola otras fallas como dejar caer la placa desde una altura suficiente pueden resultar en daños más graves.
Preparación de Nujol Mulls
Un método común de preparación de muestras sólidas para el análisis IR es el mulling. El principio aquí es al moler las partículas por debajo de la longitud de onda de la radiación incidente que va a estar pasando por allí debe haber dispersión limitada. Para suspender esas diminutas partículas, se utiliza un aceite, a menudo denominado Nujol. Se utilizan placas de sal transparentes al IR para sostener la muestra frente al haz con el fin de adquirir datos. Para preparar una muestra para el análisis IR utilizando una placa de sal, primero decidir qué segmento de la banda de frecuencia se debe estudiar, consulte Tabla\(\PageIndex{1}\) para los materiales más adecuados para la muestra. La figura\(\PageIndex{2}\) muestra los materiales necesarios para la preparación de un mull.

La preparación del mull se realiza tomando una pequeña porción de muestra y agregando aproximadamente 10% del volumen de muestra que vale el valor del aceite y moliéndolo en un mortero de ágata como se demuestra en la Figura\(\PageIndex{3}\). El mull resultante debe ser transparente sin partículas visibles.

Otro método consiste en disolver el sólido en un disolvente y dejar que se seque en la mano de mortero de ágata. Si se utiliza este método, asegúrese de que todo el disolvente se haya evaporado ya que las bandas de disolvente aparecerán en el espectro. Un poco de calentamiento suave puede ayudar a este proceso. Este método crea partículas muy finas que son de un tamaño relativamente consistente. Después de la adición del aceite puede ser necesario mezclar más (o moler).
Las placas deben almacenarse en un desecador para evitar la erosión por la humedad atmosférica y deben aparecer más o menos transparentes. Algunos materiales como el silicio no lo harán, sin embargo. Enjuague suavemente las placas con hexanos para lavar cualquier material residual de las placas. Retirar las placas del desecador y limpiarlas debe seguir la preparación del mull para mantener la integridad de las placas de sal. Por supuesto, si la placa no es soluble en agua entonces sigue siendo una buena idea solo para evitar la amenaza de trauma mecánico o un chorro de acetona extraviado de una botella de lavado.
Una vez preparada la mezcla, añadir una gota a una placa IR (Figura\(\PageIndex{4}\)), colocar la segunda placa encima de la gota y darle un cuarto de vuelta para recubrir uniformemente la superficie de la placa como se ve en la Figura\(\PageIndex{5}\). Colóquelo en el espectrómetro y adquiera los datos deseados.
Maneje siempre con guantes y preferiblemente lejos de cualquier lavamanos, grifos u otras fuentes de agua corriente o rociado.

Los espectros adquiridos por este método tendrán fuertes bandas de absorción C-H a lo largo de varios rangos 3,000 — 2,800 cm -1 y 1,500 — 1,300 cm -1 y pueden oscurecer la señal.
La limpieza de la placa se realiza como se mencionó anteriormente con hexanos o cloroformo se puede realizar fácilmente enjuagando y dejándolos secar en la campana. Vuelva a colocar las placas de sal en el desecador lo antes posible para evitar daños. Es muy recomendable pulir las placas después de su uso, no deben ser visibles rasguños, empañamientos o picaduras en la cara de la placa. Las astillas, siempre y cuando no crucen el centro de la placa son sobrevivibles pero no deseadas. Las muestras de placas de sal dañadas en la Figura\(\PageIndex{6}\) muestran problemas comunes asociados con el uso o potencialmente mal manejo. El enturbiamiento, y hasta cierto punto, los arañazos se pueden pulir con un rojo de hierro. Las áreas donde se altera la celosía cristalina debajo de la superficie son imposibles de fijar y las astillas no se pueden volver a unir.

FIGURA\(\PageIndex{6}\) Una serie de placas que indican diversas formas de daño físico con una comparación con una buena placa (Copyright: Colorado University-Boulder).
Preparación de Pellets
En un método alternativo, esta técnica es en la misma línea del mull de nujol excepto que en lugar de que el medio de suspensión sea aceite mineral, el medio de suspensión es una sal. El sólido se muele en polvo fino con un mortero de ágata y mano de mortero con una cantidad de la sal de suspensión. La preparación de pellets con diamante para el agente de suspensión es algo poco aconsejable considerando la gran dureza de la sustancia. En términos generales, se utiliza una cantidad de KBr o CSi para este método ya que ambas son sales blandas. Se pueden utilizar dos enfoques para preparar pellets, uno es algo más caro pero ambos suelen dar resultados decentes.
El primer método es el uso de una prensa. La sal se coloca en un soporte cilíndrico y se presiona junto con un ariete como el que se ve en (Figura\(\PageIndex{7}\)). Posteriormente, el pellet, en el soporte, se coloca en el instrumento y se adquieren los espectros.

Un método alternativo y más económico requiere el uso de una tuerca hexagonal grande con un diámetro interior de 0.5 pulgadas, dos pernos y dos llaves como el kit que se ve en la Figura\(\PageIndex{8}\). Las instrucciones paso a paso para cargar y usar la prensa siguen:
- Atornille uno de los pernos en la tuerca aproximadamente a la mitad del camino.
- Coloque la mezcla de pellets de sal en la otra abertura de la tuerca y nivele golpeando el conjunto en una encimera.
- Atornille el segundo perno y coloque el conjunto de lado con los pernos paralelos a la encimera. Coloca una de las llaves en el perno del lado derecho con el mango apuntando hacia ti mismo.
- Toma la segunda llave y colócala sobre el otro perno para que se adhiera con un ángulo desde la mesa de unos 45 grados.
- El segundo perno se aprieta con un peso corporal y se deja reposar durante varios minutos. Posteriormente, se retiran los pernos y se coloca la muestra en el instrumento.

Algunas prensas de pellets también tienen una púa de vacío como la que se ve en (Figura\(\PageIndex{8}\). Si su prensa de pellets tiene uno de estos, considere usarlo ya que ayudará a eliminar el aire del pellet de sal a medida que se presiona. Esto asegura un pellet más uniforme y elimina las absorbancias en el espectro recolectado debido al aire atrapado en el pellet.
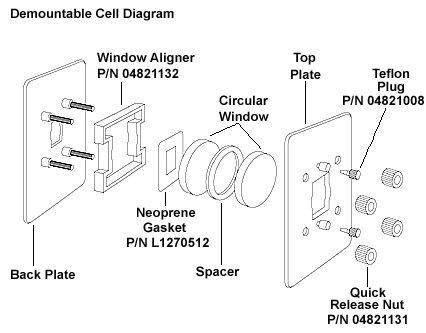
Preparación de Células de Solución
Las células de solución (Figura\(\PageIndex{9}\)) son una manera práctica de adquirir espectros infrarrojos de compuestos en solución y son particularmente útiles para monitorear reacciones.

Una celda de película delgada consiste en dos placas de sal con un espacio muy delgado entre ellas (Figura\(\PageIndex{10}\)). Dos canales permiten que el líquido sea inyectado y posteriormente eliminado. Las ventanas de estas celdas pueden estar hechas de una variedad de materiales ópticos IR. Uno particularmente útil para soluciones a base de agua es CaF 2 ya que no es soluble en agua.
La limpieza de estas células se puede realizar retirando la solución, enjuagando con disolvente fresco y retirando suavemente el disolvente mediante jeringa. No sople aire ni nitrógeno a través de los puertos ya que esto puede causar deformación mecánica en la ventana de sal si la presión es lo suficientemente alta.
Efectos de Solventes Deuterados
Uno de los otros aspectos del IR en fase de solución es que el disolvente utilizado en la célula tiene un espectro de absorción característico. En algunos casos esto se puede aliviar reemplazando el solvente por su hermano deuterado. El beneficio aquí es que los enlaces C-H son ahora enlaces C-D y tienen frecuencias vibracionales más bajas. Compilado en la Figura\(\PageIndex{11}\) es un conjunto de solventes comunes.

Este efecto tiene numerosos beneficios y a menudo se aplica para determinar qué vibraciones corresponden a qué enlace en una muestra molecular dada. Esto a menudo se logra usando reactivos “pesados” etiquetados isotópicamente como los que contienen 2 H, 15 N, 18 O o 13 C.
Solución de problemas básicos
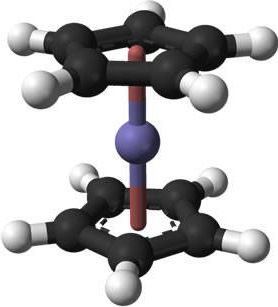
Existen numerosos problemas que pueden surgir de muestras mal preparadas, esta sección pasará por algunos de los problemas comunes y cómo corregirlos. Para esta demostración, se utilizarán espectros de ferroceno. La estructura molecular y una fotografía del compuesto organometálico de colores brillantes se muestran en la Figura\(\PageIndex{12}\) y Figura\(\PageIndex{13}\).

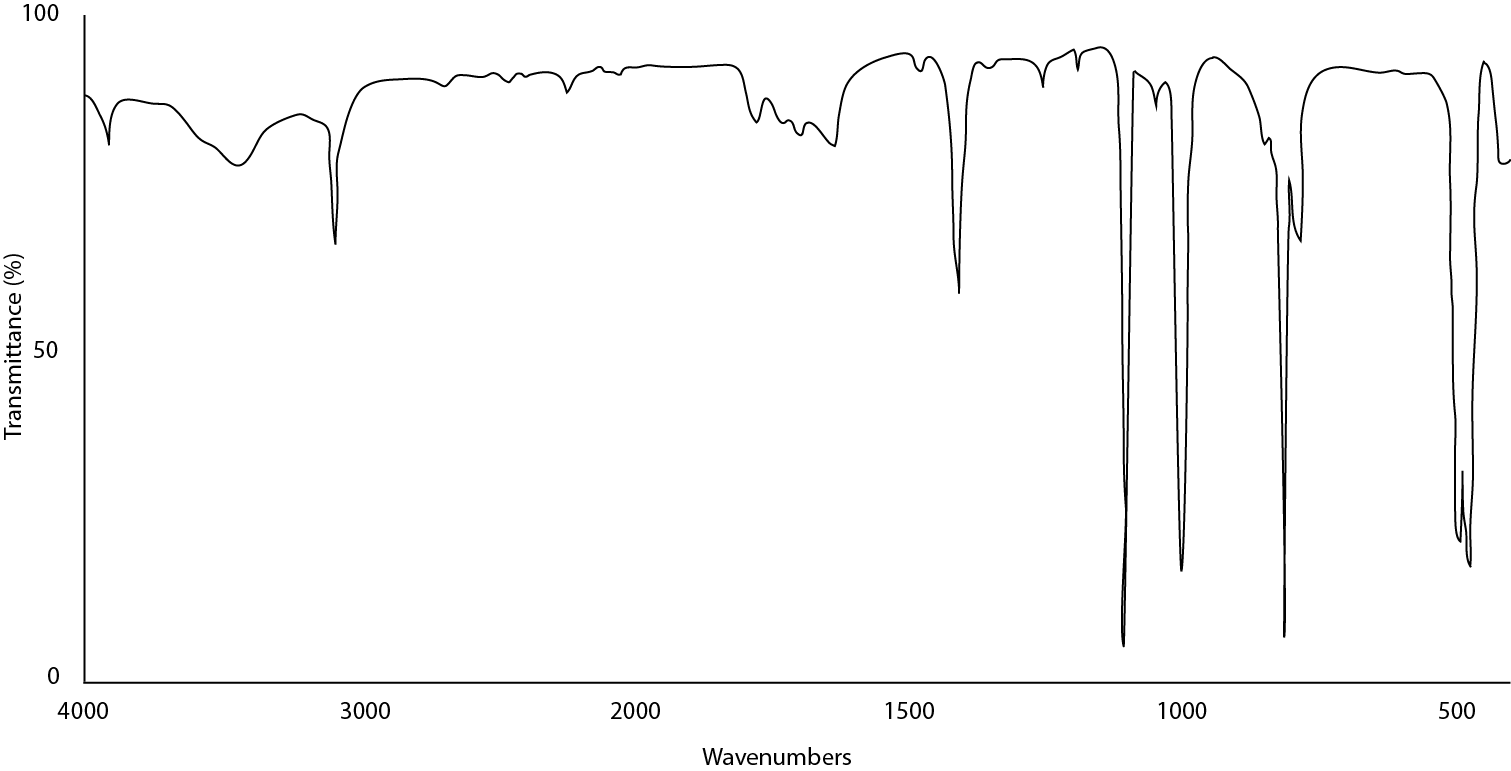
La figura\(\PageIndex{14}\) ilustra cómo se ve una buena muestra de ferroceno preparada en un pellet de KBr. Los picos están bien definidos y afilados. Ningún pico se aplana a 0% de transmitancia y la dispersión de Christiansen no es evidente en la línea base.
La figura\(\PageIndex{15}\) ilustra una muestra con algunos picos con intensidades que están saturadas y pierden resolución haciendo difícil la recolección de picos. Para corregir este problema, raspe parte de la muestra de la placa de sal con una espátula de goma y vuelva a colocar la placa opuesta. Al aplicar una capa de muestra más delgada se puede mejorar la resolución de vibraciones fuertemente absorbentes.
La figura\(\PageIndex{16}\) ilustra una muestra en la que se agregó demasiado aceite mineral al mull para que los enlaces C-H sean mucho más intensos que la muestra real. Esto se puede remediar retirando la muestra de la placa, moliendo más muestra y agregando una cantidad menor de la mezcla a la placa. Otra forma posible de hacer esto es si la muestra es insoluble en hexanos, agregar un poco al triturador y macerar la mezcla de hexano-aceite para dejar una muestra sólida seca. Aplique una pequeña porción de aceite y vuelva a colocar.
La figura\(\PageIndex{17}\) ilustra el resultado de que las partículas son demasiado grandes y dispersan la luz. Para remediar esto, retire el mull y muela más o bien use la técnica de deposición de solvente descrita anteriormente.
Modos de vibración IR característicos para compuestos hidrocarbonados
| Grupo funcional | Modo | Rango de número de onda (cm -1) |
|---|---|---|
| CH 3 | Estiramiento asimétrico | 2962±10 |
| CH 3 | Estiramiento simétrico | 2872±10 |
| CH 3 | Curva asimétrica | 1460±10 |
| CH 3 | Curva simétrica (modo paraguas) | 1375±10 |
| CH 2 | Estiramiento asimétrico | 2926±10 |
| CH 2 | Estiramiento simétrico | 2855±10 |
| CH 2 | Tijeras | 1455±10 |
| CH 2 | Rock | 720±10 |
| CH | Estiramiento | ~2900 (débil) |
| CH | Curva | ~1350 (débil) |
| Sustitución | Estiramiento C-H (cm -1) | Estiramiento C=C (cm -1) | Curva fuera del plano (cm -1) |
|---|---|---|---|
| Vinil | 3090-3075 | 1660-1630 | 900±5, 910±5 |
| Venilidina | 3090-3075 | 1660-1630 | 890±5 |
| Cis | 3050-3000 | 1660-1630 | 690±10 |
| Trans | 3050-3000 | 1680-1665 | 965±5 |
| Tri-sustituido | 3050-3000 | 1680-1665 | 815±25 |
| Tetra-sustituido | - | 1680-1665 | - |
| Sustitución | Estiramiento C-H (cm -1) | Estiramiento C=C (cm -1) | C-H wag (cm -1) |
|---|---|---|---|
| Mono-sustituido | 3350-3250 | 2140-2100 | 700-600 |
| Di-sustituido | - | 2260-2190 | - |
| Sustitución | Flexión C-H fuera del plano | Curva de anillo (cm -1) |
|---|---|---|
| Mono | 770-710 | 690±10 |
| Orto | 810-750 | - |
| Meta | 770-735 | 690±10 |
| Para | 860-790 | - |
| Vibración | Número de onda (cm -1) |
|---|---|
| CH 3 estiramiento simétrico | 2925±5 |
| Tono de curva CH 3 | 2865±5 |
Espectroscopia infrarroja por transformada de Fourier de complejos de ligandos metálicos
El rango infrarrojo (IR) del espectro electromagnético generalmente se divide en tres regiones:
- El infrarrojo lejano siempre se usa para espectroscopía rotacional, con rango de número de onda 400 — 10 cm −1 y menor energía.
- El infrarrojo medio es adecuado para una detección de las vibraciones fundamentales y la estructura rotacional-vibracional asociada con el rango de frecuencia de aproximadamente 4000 — 400 cm −1.
- El infrarrojo cercano con mayor energía y rango de número de onda 14000 — 4000 cm −1, puede excitar sobretono o vibraciones armónicas más altas.
Para la teoría clásica de interacción material ligero, si una molécula puede interactuar con un campo electromagnético y absorber un fotón de cierta frecuencia, el dipolo transitorio del grupo funcional molecular debe oscilar a esa frecuencia. Correspondientemente, este momento dipolar de transición debe ser un valor distinto de cero, sin embargo, alguna vibración especial puede ser IR inactiva para el movimiento de estiramiento de una molécula diatómica homonuclear y las vibraciones no afectan el momento dipolar de la molécula (por ejemplo, N 2).
Descripción Mecánica de las Vibraciones de Moléculas Poliatómicas
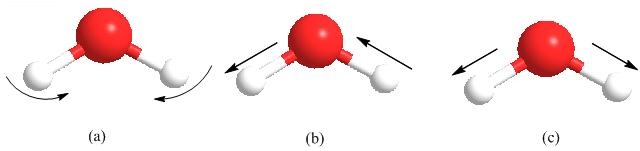
Una molécula puede vibrar de muchas maneras, y cada forma se llama un “modo vibracional”. Si una molécula tiene átomos de N, las moléculas lineales tienen 3N-5 grados de modos vibracionales mientras que las moléculas no lineales tienen 3N-6 grados de modos vibracionales. Tomemos H 2 O por ejemplo; una sola molécula de H 2 O tiene modo de flexión O-H (Figura\(\PageIndex{18}\) a), modo de estiramiento antisimétrico (Figura\(\PageIndex{18}\) b) y modo de estiramiento simétrico (Figura\(\PageIndex{18}\) c).
Si una molécula diatómica tiene una vibración armónica con la energía,\ ref {1}, donde n+ 1/2 con n = 0, 1, 2...). El movimiento de los átomos puede ser determinado por la ecuación de fuerza,\ ref {2}, donde k es la constante de fuerza). La frecuencia de vibración puede ser descrita por\ ref {3}. En el que m es realmente la masa reducida (m rojo o μ), que se determina a partir de la masa m 1 y m 2 de los dos átomos,\ ref {4}.
\[ E_{n} \ =\ -hv \label{1} \]
\[ F \ =\ -kx \label{2} \]
\[ \omega \ =\ (k/m)^{1/2} \label{3} \]
\[ m_{red} \ =\ \mu \ =\ \frac{m_{1} m_{2}}{m_{1}\ +\ m_{2} } \label{4} \]
Principio de Bandas de Absorción
En el espectro IR, la información de absorción generalmente se presenta en forma de número de onda e intensidad de absorción o porcentaje de transmitancia. El espectro generalmente muestra el número de onda (cm -1) como el eje x y la intensidad de absorción o porcentaje de transmitancia como el eje y.
La transmitancia, “T”, es la relación entre la potencia radiante transmitida por la muestra (I) y la potencia radiante incidente en la muestra (I 0). La absorbancia (A) es el logaritmo a la base 10 del recíproco de la transmitancia (T). La intensidad de absorción de la vibración molecular puede ser determinada por la Ley Lambert-Beer,\ label {5}. En esta ecuación, los espectros de transmitancia varían de 0 a 100%, y puede proporcionar un claro contraste entre intensidades de bandas fuertes y débiles. La absorbancia varía de infinito a cero. La absorción de moléculas puede ser determinada por varios componentes. En la ecuación de absorción, ε se denomina coeficiente de extinción molar, el cual se relaciona con el comportamiento de la molécula en sí, principalmente el momento dipolar de transición, c es la concentración de la muestra, y l es la longitud de la muestra. El ancho de línea puede ser determinado por la interacción con el entorno.
\[ A\ =\ log(1/T) \ =\ -log(I/I_{0} )\ =\ \varepsilon c l \label{5} \]
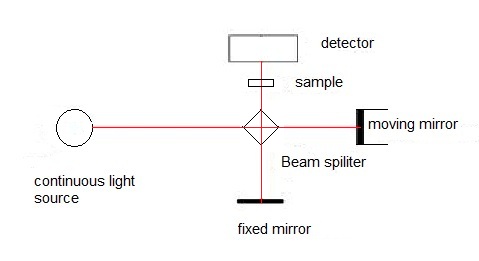
El espectrómetro de infrarrojos
Como se muestra en la Figura\(\PageIndex{19}\), hay principalmente cuatro partes para el espectrómetro infrarrojo de transformada de Fourier (FTIR):
- Fuente de luz. La energía infrarroja se emite desde una fuente de cuerpo negro brillante como radiaciones continuas.
- Interferómetro. Contiene el interferómetro, el divisor de haz, el espejo fijo y el espejo móvil. El divisor de haz toma el haz infrarrojo entrante y lo divide en dos haces ópticos. Un haz se refleja en el espejo fijo. El otro haz se refleja en el espejo móvil que se mueve una distancia muy corta. Después de que los haces divididos se reflejan desde los dos espejos, se encuentran de nuevo en el divisor de haz. Por lo tanto, se genera un patrón de interferencia por los cambios en la posición relativa del espejo móvil al espejo fijo. Luego, el haz resultante pasa a través de la muestra y finalmente se enfoca en el detector.
- Compartimento para muestras. Es el lugar donde se transmite el haz a través de la muestra. En el compartimento muestral se absorben frecuencias específicas de energía.
- Detector. El haz finalmente pasa al detector para la medición final. Los dos detectores más populares para un espectrómetro FTIR son el sulfato de triglicina deuterado (detector piroeléctrico) y el telururo de mercurio cadmio (detector de fotones o cuánticos). La señal medida se envía a la computadora donde tiene lugar la transformación de Fourier.
Una aplicación típica: la detección de complejos de ligandos metálicos
Algunos picos de absorción general para tipos comunes de grupos funcionales
Es bien sabido que todas las moléculas químicas tienen distintas regiones de absorción en el espectro IR. En el cuadro se\(\PageIndex{7}\) muestran las frecuencias de absorción de tipos comunes de grupos funcionales. Para la evaluación sistemática, el espectro IR se divide comúnmente en algunas subregiones.
- En la región de 4000 - 2000 cm —1, la aparición de bandas de absorción generalmente proviene del estiramiento de vibraciones entre el hidrógeno y otros átomos. Las frecuencias de estiramiento O-H y N-H oscilan entre 3700 - 3000 cm —1. Si se forma un enlace de hidrógeno entre O-H y otro grupo, generalmente causó el ensanchamiento de la forma de la línea de pico y el desplazamiento a frecuencias más bajas. Las bandas de estiramiento C-H ocurren en la región de 3300 - 2800 cm —1. El C-H acetilénico presenta una fuerte absorción alrededor de 3300 cm —1. Alqueno y las vibraciones aromáticas de estiramiento C-H absorben a 3200-3000 cm —1. Generalmente, la frecuencia de estiramiento vibracional asimétrico del alqueno C-H es de alrededor de 3150 cm -1, y la frecuencia de estiramiento vibracional simétrico está entre 3100 cm -1 y 3000 cm -1. Las bandas de estiramiento C-H alifáticas saturadas oscilan entre 3000 - 2850 cm —1, con intensidades de absorción que son proporcionales al número de enlaces C-H. Los aldehídos a menudo muestran dos bandas de absorción de estiramiento C-H afiladas a 2900 - 2700 cm —1. Sin embargo, en solución acuosa, el estiramiento vibratorio C-H es mucho menor que en la solución no polar. Esto significa que la solución de polaridad fuerte puede reducir en gran medida el momento dipolar de transición de la vibración C-H.
- Además, las frecuencias de vibración de estiramiento entre hidrógeno y otros heteroátomos están entre 2600 - 2000cm -1, por ejemplo, S-H a 2600 - 2550 cm —1, P-H a 2440 - 2275 cm —1, Si-H a 2250 - 2100 cm —1.
- Las bandas de absorción en la región 2300 - 1850 cm —1 generalmente se presentan únicamente a partir de enlaces triples, como C≡ C a 2260 - 2100 cm —1, C≡ N a 2260 - 2000 cm —1, sales de diazonio —N=N a aproximadamente 2260 cm —1, alenos C=C=C a 2000 - 1900 cm —1. Los picos de estos grupos son todos tienen fuertes intensidades de absorción. La región 1950 - 1450 cm —1 representa el estiramiento vibracional de grupos funcionales de doble enlace.
- La mayoría de las bandas de estiramiento de carbonilo C=O oscilan entre 1870 y 1550 cm —1, y las intensidades de los picos son de media a fuerte. La conjugación, el tamaño del anillo, los enlaces de hidrógeno y los efectos estéricos y electrónicos pueden conducir a cambios significativos en las frecuencias de absorción. Además, si el carbonilo se une con un grupo aceptor de electrones, como cloruros de ácido y anhídridos de ácido, daría lugar a bandas IR a 1850 - 1750 cm —1. Las cetonas suelen mostrar bandas de estiramiento a 1715 cm -1.
- Ninguno alifático conjugado C=C y C=N tiene bandas de absorción a 1690 - 1620 cm —1. Además, alrededor de 1430 y 1370cm -1, hay dos picos idénticos que presentan flexión C-H.
- La región de 1300 a 910 cm —1 siempre incluye las contribuciones de los estiramientos vibracionales C-O y C-C del esqueleto, dando información estructural molecular adicional correlacionada con áreas de mayor frecuencia. Por ejemplo, el acetato de etilo no solo muestra su estiramiento carbonilo a 1750 - 1735 cm —1, sino que también exhibe sus picos de absorción idénticos a 1300 - 1000 cm —1 de la vibración del esqueleto de los estiramientos C-O y C-C.
| Grupo | Frecuencia (cm -1) | Apariencia de fuerza |
| Estiramiento C-H | 2850-3400 | Fuerte en disolvente no polar Débil en disolvente polar |
| Estiramiento O-H, estiramiento N-H | 3200-3700 | Amplio en solvente |
| Estiramiento C=N, estiramiento R-N=C=S |
2050-2300 | Mediano o fuerte |
| Estiramiento Cº (encuadernado con metal) | alrededor de 2000 | Mediano o fuerte |
| Estiramiento C=C | 2100-2260 | Débil |
| Estiramiento C=O | ca 1715 (cetona), ca 1650 (amidas) |
Fuerte |
| Estiramiento C=C | 1450-1700 | Débil a fuerte |
| Curva C-H | 1260 - 1470 | Fuerte |
| Estiramiento C-O | 1040-1300 | Mediano o fuerte |
Introducción general del complejo de ligando metálico
Los electrones metálicos se llenan en el orbital molecular de ligandos (CN, CO, etc.) para formar compuestos complejos. Como se muestra en la Figura\(\PageIndex{20}\), se puede utilizar un diagrama orbital molecular simple para CO para explicar el mecanismo de unión.
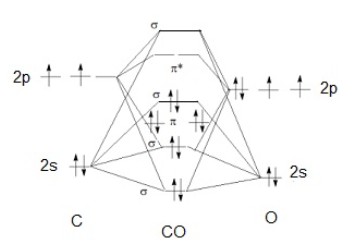
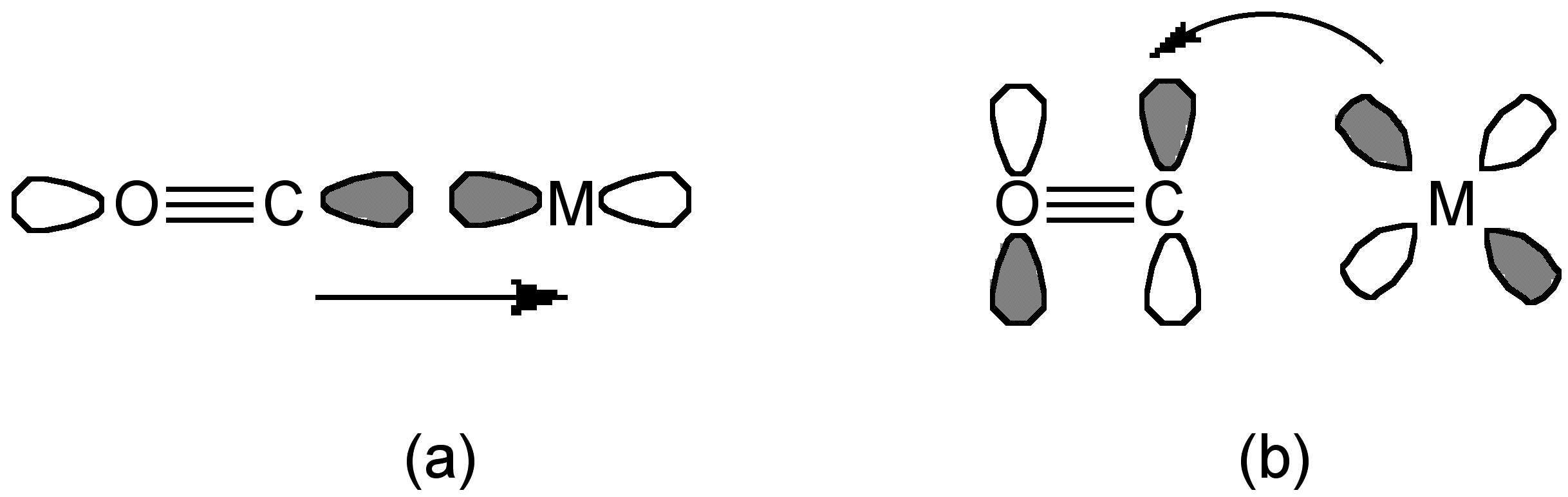
El CO y el metal pueden unirse de tres maneras:
- Donación de un par de electrones del orbital C-O σ* a un orbital metálico vacío (Figura\(\PageIndex{21}\) a).
- Donación de una órbita de metal d al orbital C-O π* para formar un enlace m-a-CO π-back (Figura\(\PageIndex{21}\) b).
- Bajo algunas condiciones, un par de electrones π de carbono puede donar en un d-orbital de metal vacío.
Algunos factores para incluir los cambios de banda y la fuerza
Aquí consideramos principalmente dos propiedades: la frecuencia de estiramiento del ligando y su intensidad de absorción. Tomemos de nuevo el ligando CO por ejemplo. El desplazamiento de frecuencia de los picos de carbonilo en el IR depende principalmente del modo de unión del CO (terminal o puente) y la densidad electrónica en el metal. La intensidad y los números de pico de las bandas de carbonilo dependen de algunos factores: números de ligandos de CO, geometría del complejo ligando metálico y resonancia fermi.
Efecto sobre la Densidad Electrónica en el Metal
Como se muestra en la Tabla\(\PageIndex{8}\), una mayor carga en el centro metálico da como resultado la disminución de la frecuencia de vibración de los estiramientos de CO Por ejemplo, [Ag (CO)] +muestran mayor frecuencia de CO que CO libre, lo que indica un fortalecimiento o
f el enlace CO. La donación σ elimina la densidad de electrones del HOMO no enlazante del CO. De la Figura, es claro que el HOMO tiene una pequeña cantidad de propiedad antiunión, por lo que la eliminación de un electrón en realidad aumenta (ligeramente) la fuerza de unión de CO. Por lo tanto, el efecto de carga y electronegatividad depende de la cantidad de unión de metal a CO π-back y de la frecuencia de estiramiento CO IR.
| d x | Complejo | Frecuencia de estiramiento de CO (cm -1) |
| CO gratis | 2143 | |
| d 10 | [Ag (CO)] + | 2204 |
| d 10 | Ni (CO) 4 | 2060 |
| d 10 | [Co (CO) 4] - | 1890 |
| d 6 | [Mn (CO) 6] + | 2090 |
| d 6 | Cr (CO) 6 | 2000 |
| d 6 | [V (CO) 6] - | 1860 |
Si la densidad de electrones en un centro metálico está aumentando, también se incrementarán más enlaces π-back al (los) ligando (s) de CO, como se muestra en la Tabla\(\PageIndex{9}\). Significa que una mayor densidad de electrones entraría en el orbital de carbonilo π* vacío y debilitaría el enlace C-O. Por lo tanto, hace que la fuerza de unión M-CO aumente y sea más similar a la de doble enlace (M=C=O).
Efecto de donación de ligadura
Algunos casos, como se muestra en la Tabla\(\PageIndex{9}\), diferentes ligandos se unirían con el mismo metal en el mismo complejo metal-ligando. Por ejemplo, si diferentes grupos de densidad de electrones se unen con Mo (CO) 3 de la misma forma, como se muestra en la Figura\(\PageIndex{22}\), las frecuencias vibracionales de CO dependerían del efecto de donación del ligando. En comparación con el grupo PPh 3, la frecuencia de estiramiento de CO a la que el complejo se une al grupo PF 3 (2090, 2055 cm -1) es mayor. Indica que la cantidad absoluta de densidad electrónica en ese metal puede tener cierto efecto sobre la capacidad de los ligandos en un metal para donar densidad electrónica al centro del metal. De ahí que pueda explicarse por el efecto de donación de Ligando. Los ligandos que son trans a un carbonilo pueden tener un gran efecto sobre la capacidad del ligando de CO para enlazarse de manera efectiva π-backlink con el metal. Por ejemplo, dos ligandos trans π-backbonding competirán parcialmente por la misma densidad de electrones d-orbital, debilitando el enlace posterior neto M-L π-backbonding del otro. Si el ligando trans es un ligando donador de π, el enlace posterior de metal libre a CO puede aumentar la fuerza del enlace M-CO (más carácter M=C=O). Es bien sabido que la piridina y las aminas no son esos donadores p fuertes. Sin embargo, son incluso peores ligandos de unión por retroceso π. Entonces el CO es realmente fácil para la donación π-back sin ninguna competencia. Por lo tanto, reduce naturalmente las frecuencias de estiramiento de CO IR en complejos de carbonilo metálico para el efecto de donación de ligandos.
| Complejo de ligando metálico | Frecuencia de Estiramiento CO (cm -1) |
|---|---|
| Mo (CO) 3 (PF 3) 3 | 2090, 2055 |
| Mo (CO) 3 [P (OMe) 3] 3 | 1977, 1888 |
| Mo (CO) 3 (PPh 3) 3 | 1934, 1835 |
| Mo (CO) 3 (NCCH 3) 3 | 1915, 1783 |
| Mo (CO) 3 (piridina) 3 | 1888, 1746 |
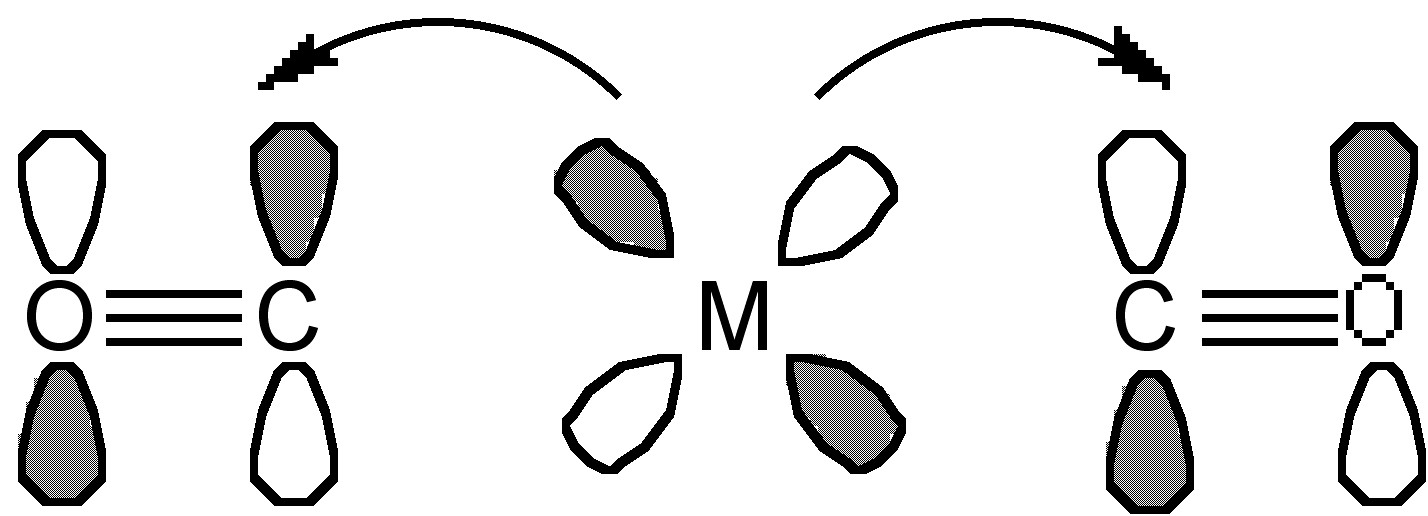
Efectos de Geometría
En algunos casos, el complejo metal-ligando puede formar geometría no solo terminal sino también puente. Como se muestra en la Figura\(\PageIndex{23}\), en el compuesto Fe 2 (CO) 7 (dipy), el CO puede actuar como ligando puente. La evidencia de un modo puente de coordinación se puede obtener fácilmente a través de espectroscopía IR. Todos los átomos metálicos puenteados por un carbonilo pueden donar densidad electrónica al orbital π* del CO y debilitar el enlace CO, disminuyendo la frecuencia de vibración del CO. En este ejemplo, la frecuencia de CO en terminal es de alrededor de 2080 cm -1, y en puente, se desplaza a alrededor de 1850 cm -1.
Detección de bomba-sonda de vida vibracional del grupo funcional molecular
La dinámica del grupo funcional molecular juega un papel importante durante un proceso químico, la formación y ruptura de enlaces químicos, la transferencia de energía y otras dinámicas ocurren dentro del dominio de los picosegundos. Es muy difícil estudiar procesos tan rápidos directamente, durante décadas los científicos solo pueden aprender de cálculos teóricos, careciendo de métodos experimentales.
Sin embargo, con el desarrollo de láser pulsado ultracorto permiten el estudio experimental de la dinámica de grupos funcionales moleculares. Con tecnologías láser ultrarrápidas, las personas desarrollan una serie de métodos de medición, entre los cuales, la técnica bomba-sonda es ampliamente utilizada para estudiar la dinámica de grupos funcionales moleculares. Aquí nos concentramos en cómo usar el experimento bomba-sonda para medir la vida de la vibración del grupo funcional. Se introducirá el principio, la configuración experimental y el análisis de datos.
Principios de la Técnica de Bomba-Sonda
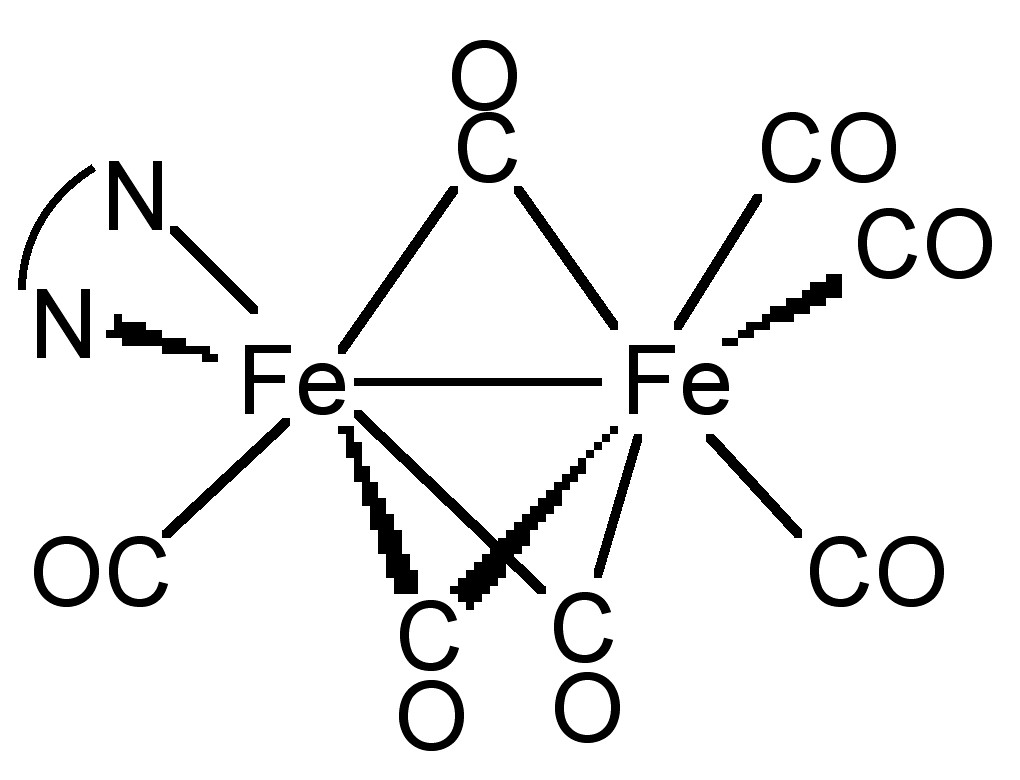
Por cada grupo funcional dentro de una molécula, como el triple enlace C=N en selenocianato de fenilo (C 6 H 5 SecN) o el enlace sencillo C-D en cloroformo deuterado (DCCl 3), tienen un modo vibracional infrarrojo individual y niveles de energía asociados. Para un sistema típico de 3 niveles (Figura\(\PageIndex{24}\), tanto la transición 0 a 1 como la de 1 a 2 están cerca de la frecuencia del pulso de la sonda (no necesariamente necesitan tener exactamente la misma frecuencia).
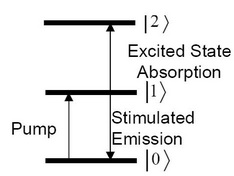
En un experimento bomba-sonda, utilizamos la geometría como se muestra en la Figura\(\PageIndex{25}\). Dos haces láser sincronizados, uno de los cuales se llama haz de bomba (E pu) mientras que el otro haz sonda (E pr). Hay un retraso en el tiempo entre cada pulso. Los pulsos de láser golpean la muestra, la intensidad del láser ultrarrápido (fs o ps) es lo suficientemente fuerte como para generar polarización de 3 rd orden y producir una señal de respuesta óptica de 3 rd orden que se usa para dar información dinámica de grupos de función molecular. Para las señales de respuesta total tenemos\ label {6}, donde µ 10 µ 21 son momento dipolar de transición y E 0, E 1 y E2 son las energías de los tres niveles, y t 3 es el retardo de tiempo entre la bomba y el haz sonda. Se varía el retardo t3 y se mide la intensidad de la señal de respuesta. El tiempo de vida de vibración del grupo funcional se determina a partir de los datos.
\[ S \ =\ 4 \mu _{10} ^{4} e^{ -i(E_{1} - E_{0} ) t3/h - \Gamma t3} \label{6} \]
Configuración Experimental Típica
El diseño óptico de una configuración típica de bomba-sonda se muestra esquemáticamente en la Figura\(\PageIndex{26}\). En la configuración, la salida del oscilador (500 mW a una tasa de repetición de 77 MHz, ancho de banda de 40 nm centrado a 800 nm) se divide en dos haces (relación de potencia 1:4). De esto, el 20% de la potencia consiste en sembrar un amplificador de femtosegundo (fs) cuya salida es de 40 pulsos fs centrados a 800 nm con una potencia de ~3.4 W a una tasa de repetición de 1 kHz. El resto (80%) de la semilla pasa por un filtro pasabanda centrado a 797.5nm con un ancho de 0.40 nm para sembrar un amplificador de picosegundos (ps). La potencia de la semilla estirada antes de ingresar a la cavidad del amplificador ps es de solo ~3 mW. La salida del amplificador ps es pulsos de 1ps centrados a 800 nm con un ancho de banda de ~0.6 nm. La potencia de la salida del amplificador ps es de ~3 W. El amplificador fs es entonces para bombear un amplificador paramétrico óptico (OPA) que produce ~100 fs pulsos IR con ancho de banda de ~200 cm -1 que es sintonizable de 900 a 4000 cm -1. La potencia de los pulsos fs IR es de 7~40 mW, dependiendo de las frecuencias. El amplificador ps es para bombear un ps OPA que produce ~900 fs pulsos IR con ancho de banda de ~21 cm -1, sintonizable de 900 - 4000 cm -1. La potencia de los pulsos fs IR es de 10 ~ 40 mW, dependiendo de las frecuencias.
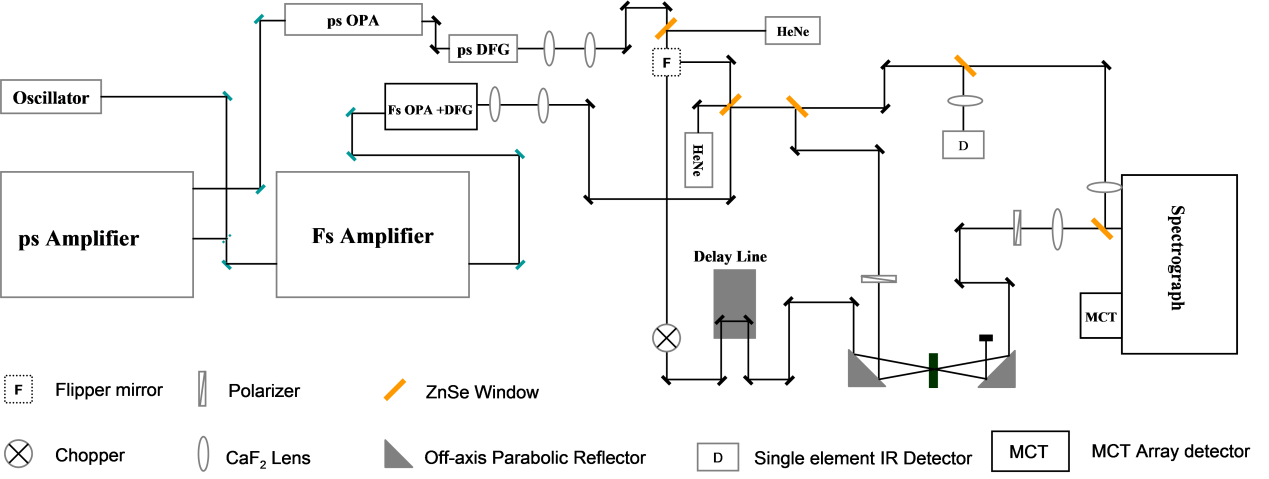
En una configuración típica de bomba-sonda, el haz ps IR se colima y se usa como el haz de bomba. Aproximadamente el 1% de la salida fs IR OPA se utiliza como el haz sonda cuya intensidad es modificada adicionalmente por un polarizador colocado antes de la muestra. Otro polarizador se coloca después de la muestra y antes del espectrógrafo para seleccionar diferentes polarizaciones de la señal. Luego, la señal se envía a un espectrógrafo para resolver la frecuencia y se detecta con un detector de matriz dual de telururo de mercurio cadmio (MCT). El uso de un pulso de bomba (femtosegundo, banda ancha) y un pulso de sonda (picosegundos, banda estrecha), escaneando el tiempo de retardo y leyendo los datos del espectrómetro, darán la vida útil del grupo funcional. La bomba de banda ancha y el espectrómetro descritos aquí son para recolectar múltiples grupos de combinación bomba-sonda.
Análisis de datos
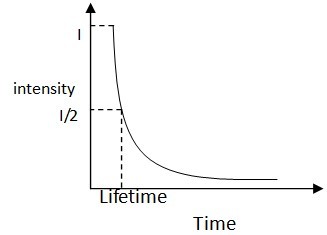
Para una curva típica bomba-sonda mostrada en la Figura, el tiempo de\(\PageIndex{27}\) vida t se define como el valor de tiempo correspondiente a la media intensidad como tiempo cero.
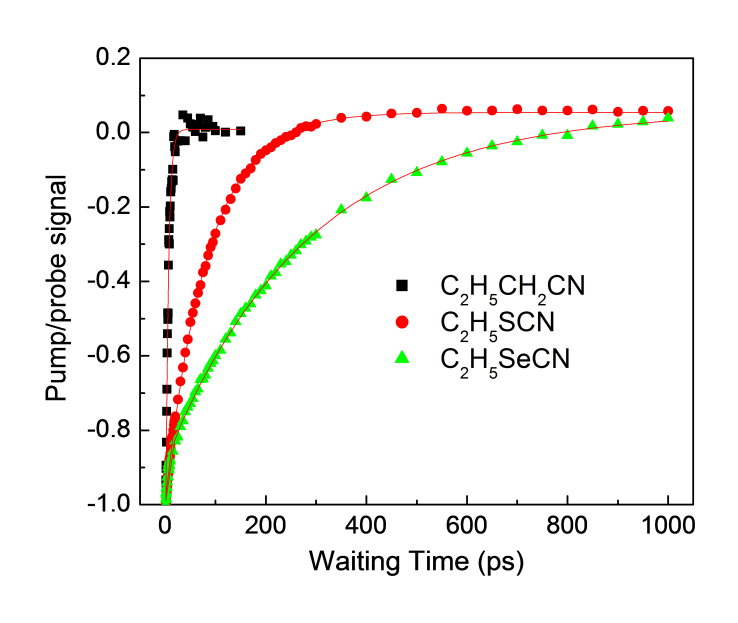
En la tabla se\(\PageIndex{10}\) muestran los datos bomba-sonda del triple enlace C≡ N en una serie de compuestos de ciano aromáticos: cianuro de n-propilo (C 3 H 7 CN), tiocianato de etilo (C 2 H 5 SCN) y selenocianato de etilo (C 2 H 5 SecN) para los que se muestra el ν C = N para cada compuesto (medido en solución CCl 4) es 2252 cm -1), 2156 cm -1 y ~2155 cm -1, respectivamente.
| Retraso (ps) | C 3 H 7 CN | C 2 H 5 SCN | C 2 H 5 SECN |
|---|---|---|---|
| 0 | -0.00695 | -0.10918 | -0.06901 |
| 0.1 | -0.0074 | -0.10797 | -0.07093 |
| 0.2 | -0.00761 | -0.1071 | -0.07247 |
| 0.3 | -0.00768 | -0.10545 | -0.07346 |
| 0.4 | -0.0076 | -0.10487 | -0.07429 |
| 0.5 | -0.00778 | -0.10287 | -0.07282 |
| 0.6 | -0.00782 | -0.10286 | -0.07235 |
| 0.7 | -0.00803 | -0.10222 | -0.07089 |
| 0.8 | -0.00764 | -0.10182 | -0.07073 |
| 0.9 | -0.00776 | -0.10143 | -0.06861 |
| 1 | -0.00781 | -0.10099 | -0.06867 |
| 1.1 | -0.00745 | -0.10013 | -0.06796 |
| 1.2 | -0.00702 | -0.10066 | -0.06773 |
| 1.3 | -0.00703 | -0.0989 | -0.0676 |
| 1.4 | -0.00676 | -0.0995 | -0.06638 |
| 1.5 | -0.00681 | -0.09757 | -0.06691 |
| 1.6 | -0.00639 | -0.09758 | -0.06696 |
| 1.7 | -0.00644 | -0.09717 | -0.06583 |
| 1.8 | -0.00619 | -0.09741 | -0.06598 |
| 1.9 | -0.00613 | -0.09723 | -0.06507 |
| 2 | -0.0066 | -0.0962 | -0.06477 |
| 2.5 | -0.00574 | -0.09546 | -0.0639 |
| 3 | -0.0052 | -0.09453 | -0.06382 |
| 3.5 | -0.0482 | -0.09353 | -0.06389 |
| 4 | -0.0042 | -0.09294 | -0.06287 |
| 4.5 | -0.00387 | -0.09224 | -0.06197 |
| 5 | -0.00351 | -0.09009 | -0.06189 |
| 5.5 | -0.00362 | -0.09084 | -0.06188 |
| 6 | -0.00352 | -0.08938 | -0.06021 |
| 6.5 | -0.00269 | -0.08843 | -0.06028 |
| 7 | -0.00225 | -0.08788 | -0.05961 |
| 7.5 | -0.00231 | -0.08694 | -0.06065 |
| 8 | -0.00206 | -0.08598 | -0.05963 |
| 8.5 | -0.00233 | -0.08552 | -0.05993 |
| 9 | -0.00177 | -0.08503 | -0.05902 |
| 9.5 | -0.00186 | -0.08508 | -0.05878 |
| 10 | -0.00167 | -0.0842 | -0.0591 |
| 11 | -0.00143 | -0.08295 | -0.05734 |
Se muestra una gráfica de intensidad versus tiempo para los datos de TABLE Figura\(\PageIndex{28}\). A partir de estas curvas se pueden determinar los tiempos de vida de estiramiento de C≡ N para C 3 H 7 CN, C 2 H 5 SCN y C 2 H 5 SecN como ~5.5 ps, ~84 ps y ~282 ps, respectivamente.
De lo que se muestra arriba, el método bomba-sonda se utiliza en la detección de vidas vibratorias de C=N en diferentes productos químicos. Una medición solo toma varios segundos para obtener todos los datos y la vida útil, lo que demuestra que el método bomba-sonda es una forma poderosa de medir la vida útil de la vibración del grupo funcional.
Reflectancia total atenuada - Espectroscopia infrarroja por transformada de Fourier
Reflectancia total atenuada - La espectroscopia infrarroja por transformada de Fourier (ATR-FTIR) es un método físico de análisis composicional que se basa en la espectroscopia FTIR de transmisión tradicional para minimizar la preparación de muestras y optimizar la reproducibilidad. Las muestras de fase condensada de índice de refracción relativamente bajo se colocan en estrecho contacto con un cristal de alto índice de refracción y se puede recolectar el espectro de absorción infrarroja (IR) de la muestra. Basado en la reflexión interna total, los espectros de absorción de ATR se asemejan a los de transmisión FTIR. Para obtener más información sobre la espectroscopia IR de transmisión (FTIR) consulte la sección más arriba en esta página titulada Espectroscopia infrarroja por transformada de Fourier de complejos de ligandos metálicos.
Propuesta públicamente por primera vez en 1959 por Jacques Fahrenfort de los laboratorios Royal Dutch Shell en Ámsterdam, la espectroscopia ATR IR fue descrita como una técnica para medir eficazmente materiales de fase condensada de absorción débil. En el primer artículo de Fahrenfort que describe la técnica, publicado en 1961, utilizó un cristal ATR hemicilindrico (ver Condiciones Experimentales) para producir ATR de reflexión única (Figura\(\PageIndex{29}\)). La espectroscopia ATR IR tardó en ser aceptada como método de caracterización debido a preocupaciones sobre su efectividad cuantitativa y reproducibilidad. La principal preocupación es el contacto de la muestra y el cristal ATR necesario para lograr un contraste espectral decente. A finales de la década de 1980, los espectrómetros FTIR comenzaron a mejorar debido al aumento del rango dinámico, la relación señal/ruido y las computadoras más rápidas. Como resultado ATR-FTIR también comenzó a ganar tracción como una técnica espectroscópica eficiente. En estos días los accesorios ATR a menudo se fabrican para trabajar en conjunto con la mayoría de los espectrómetros FTIR, como se puede ver en la Figura\(\PageIndex{30}\).


Reflejo Interno Total
Para obtener información adicional sobre las ondas de luz y sus propiedades, consulte el módulo sobre Interferometría de Barrido Vertical (VSI) en el capítulo 10.1.
Al considerar que la luz se propaga a través de una interfaz entre dos materiales con diferentes índices de refracción, el ángulo de refracción puede ser dado por la ley de Snell,\ ref {7}, donde no se transmitirá ninguna luz incidente.
\[ \varphi _{c} \ =\ \varphi _{max} \label{7} \]
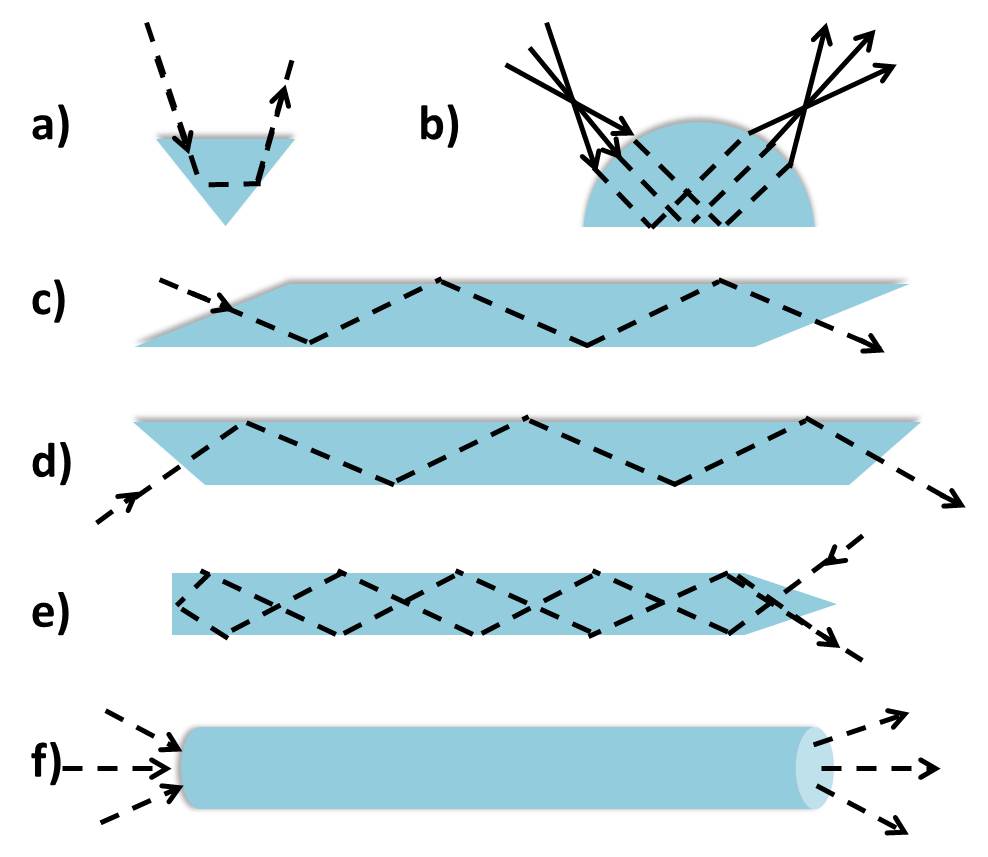
La reflectancia de la interfaz es total y siempre que la luz incide desde un medio de índice de refracción más alto sobre un medio de índice de refracción inferior, la reflexión se considera interna (a diferencia de externa en el escenario opuesto). La reflectancia interna total no experimenta pérdidas ni luz transmitida (Figura\(\PageIndex{31}\)
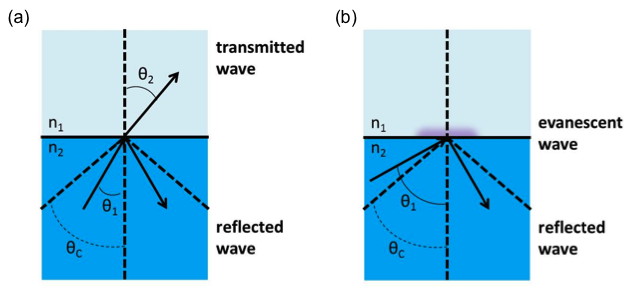
La reflexión interna supercrítica se refiere a ángulos de incidencia por encima del ángulo crítico de incidencia permitiendo reflectancia interna total. Es en este régimen angular donde solo estarán presentes las ondas incidentes y reflejadas. La onda transmitida se limita a la interfaz donde su amplitud es máxima y se humedecerá exponencialmente en el medio de índice de refracción inferior en función de la distancia. Esta onda se conoce como la onda evanescente y se extiende solo una distancia muy corta más allá de la interfaz.
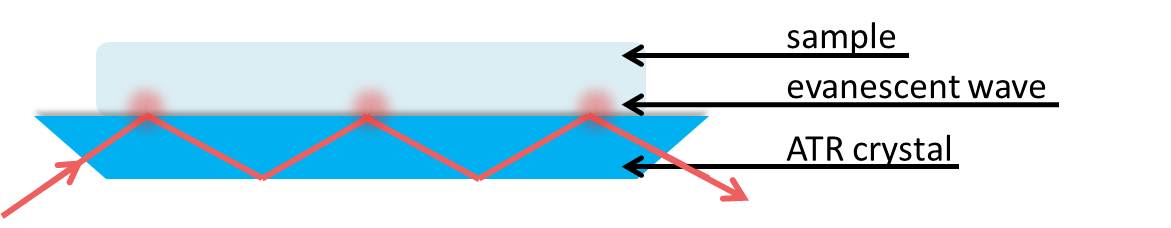
Para aplicar la reflexión interna total a la configuración experimental en ATR, considérese n 2 como el elemento de reflectancia interna o cristal ATR (el trapecio azul en la Figura\(\PageIndex{32}\))
donde n 2 es el material con mayor índice de refracción. Este debe ser un material que sea totalmente transparente a la radiación infrarroja incidente para dar un valor real para el índice de refracción. El cristal ATR también debe tener un alto índice de refracción para permitir la reflexión interna total con muchas muestras que tienen un índice de refracción n 1, donde n 1 < n 2.
Podemos considerar que la muestra es absorbente en el infrarrojo. La energía electromagnética pasará a través de la interfaz cristal/muestra y se propagará a la muestra a través de la onda evanescente. Esta pérdida de energía debe ser compensada con la luz IR incidente. Por lo tanto, la reflectancia total ya no se produce y la reflexión dentro del cristal se atenúa. Si una muestra no absorbe, la reflectancia en la interfaz no muestra atenuación. Por lo tanto, si la luz IR a una frecuencia determinada no llega al detector, la muestra debe haberla absorbido.
La profundidad de penetración de la onda evanescente dentro de la muestra es del orden de 1µm. La expresión de la profundidad de penetración se da en\ ref {8} y depende de la longitud de onda y ángulo de la luz incidente así como de los índices de refracción del cristal ATR y la muestra. La longitud efectiva de la trayectoria es el producto de la profundidad de penetración de la onda evanescente y del número de puntos que la luz IR refleja en la interfaz entre el cristal y la muestra. Esta longitud de ruta es equivalente a la longitud de ruta de una muestra en una configuración de FTIR de transmisión tradicional.
\[ d_{p} = \frac{ \lambda }{2 \pi n_{1}} (sin \omega - ( \frac{n_{1}}{n_{2}} )^{2} )^{1/2} \label{8} \]
Condiciones Experimentales
Índices de refracción de cristal ATR y muestra
Típicamente, se puede usar un accesorio ATR con un FTIR tradicional donde el haz de luz IR incidente ingresa a un cristal posicionado horizontalmente con un alto índice de refracción en el rango de 1.5 a 4, como se puede ver en la Tabla\(\PageIndex{11}\) consistirá en compuestos orgánicos, compuestos inorgánicos y polímeros que tienen índices de refracción inferiores a 2 y se pueden encontrar fácilmente en una base de datos.
| Material | Índice de Refracción (RI) | Rango espectral (cm -1) |
|---|---|---|
| Seleniuro de Zinc (ZnSe) | 2.4 | 20,000 - 650 |
| Germanio (Ge) | 4 | 5,500 - 870 |
| Zafiro (Al 2 O 3) | 1.74 | 50,000 - 2,000 |
| Diamante (C) | 2.4 | 45,000 - 2,500, 1650 - 200 |
Cristales de Reflexión Únicos y Múltiples
La ATR de reflexión múltiple fue inicialmente más popular que la ATR de reflexión simple debido a las débiles absorbancias asociadas con la ATR de reflexión única. Más reflexiones incrementaron la interacción de la onda evanescente con la muestra, lo que se creía que aumentaba la relación señal/ruido del espectro. Cuando los espectrómetros IR desarrollaron un mejor contraste espectral, el ATR de reflexión única se hizo más popular. El número de reflexiones y contraste espectral aumenta con la longitud del cristal y disminuye con el ángulo de incidencia así como el grosor. Dentro de múltiples cristales de reflexión se transmite parte de la luz y otra se refleja a medida que la luz sale del cristal, lo que da como resultado que parte de la luz regrese a través del cristal para un viaje de ida y vuelta. Por lo tanto, la luz que sale del cristal ATR contiene componentes que experimentaron diferente número de reflexiones en la interfaz cristal-muestra.
Ángulo de Incidencia
Era más común en instrumentos anteriores permitir la selección del ángulo incidente, ofreciendo a veces una selección entre 30°, 45° y 60°. En todos los casos para que se mantenga la reflexión interna total, el ángulo de incidencia debe superar el ángulo crítico e idealmente complementar el ángulo del borde del cristal para que la luz entre en un ángulo de incidencia normal. En estos días 45° es el ángulo estándar en la mayoría de las configuraciones ATR-FTIR.
Forma de Cristal ATR
En su mayor parte los cristales ATR tendrán una forma trapezoidal como se muestra en la Figura\(\PageIndex{31}\). Esta forma facilita la preparación y manejo de la muestra en la superficie del cristal al permitir que la configuración óptica se coloque debajo del cristal. Sin embargo, se pueden usar diferentes formas de cristal (Figura\(\PageIndex{33}\)) para fines particulares, ya sea para lograr múltiples reflejos o reducir el tamaño del punto. Por ejemplo, se puede usar un cristal hemisférico en un experimento de micromuestreo en el que el diámetro del haz se puede reducir sin ningún costo a la intensidad de la luz. Esto permite la medición apropiada de una pequeña muestra sin comprometer la calidad de las características espectrales resultantes.
Contacto cristal-muestra
Debido a que la longitud de la trayectoria de la onda evanescente está confinada a la interfaz entre el cristal ATR y la muestra, la muestra debe hacer contacto firme con el cristal ATR (Figura\(\PageIndex{34}\)). La muestra se asienta sobre el cristal y se puede asegurar un contacto íntimo aplicando presión sobre la muestra. Sin embargo, hay que tener en cuenta la dureza del cristal ATR. Demasiada presión puede distorsionar el cristal y afectar la reproducibilidad del espectro resultante.

El efecto de longitud de onda expresado en\ label {7} muestra un aumento en la profundidad de penetración a mayor longitud de onda. En términos de números de onda la relación se vuelve inversa. A 4000 cm -1, la penetración de la muestra es 10x menor que la penetración a 400 cm -1, lo que significa que la intensidad de los picos puede aparecer mayor a números de onda más bajos en el espectro de absorbancia en comparación con las características espectrales en un espectro FTIR de transmisión (si se realiza una corrección automatizada al ATR la configuración no está ya en su lugar).
Selección de un cristal ATR
ATR funciona eficazmente con la condición de que el índice de refracción del cristal sea de un índice de refracción mayor que la muestra. Varios cristales están disponibles para su uso y es importante seleccionar una opción apropiada para cualquier experimento dado (Tabla\(\PageIndex{11}\)).
Al seleccionar un material, es importante considerar la reactividad, la temperatura, la toxicidad, la solubilidad y la dureza.
Los primeros cristales de ATR en uso fueron KRS-5, una mezcla de bromuro y yoduro de talio, y haluros de plata. Estos materiales no están listados en la tabla porque ya no están en uso. Si bien son rentables, no son prácticas debido a su sensibilidad a la luz, suavidad e índices de refracción relativamente bajos. Además, el KRS-5 es terriblemente tóxico y se disuelve al entrar en contacto con muchos disolventes, entre ellos el agua.
En la actualidad el diamante es una opción favorable por su dureza, inercia y amplio rango espectral, pero puede no ser una opción financieramente viable para algunos experimentos. El ZnSe y el germanio son los materiales cristalinos más comunes. ZnSe tiene un precio razonable, tiene una resistencia mecánica significativa y una larga resistencia. Sin embargo, la superficie se grabará con la exposición a productos químicos en cualquier extremo de la escala de pH. Con un ácido fuerte ZnSe reaccionará para formar gas seleniuro de hidrógeno tóxico. El ZnSe también es propenso a la oxidación y se debe tener cuidado para evitar la formación de una capa absorbente de IR de SeO 2. El germanio tiene un índice de refracción más alto, lo que reduce la profundidad de penetración a 1 µm y puede ser preferible al ZnSe en aplicaciones que involucran absorciones intensas de muestra o para su uso con muestras que producen fuertes absorciones de fondo. El zafiro es físicamente robusto con un amplio rango espectral, pero tiene un índice de refracción relativamente bajo en términos de cristales ATR, lo que significa que es posible que no pueda probar tantas muestras como podría ser otro cristal.
Versatilidad de muestra
Sólidos
La versatilidad del ATR se refleja en las diversas formas y fases que una muestra puede asumir. Las muestras sólidas no necesitan comprimirse en un sedimento, dispersarse en una mezcla o disolverse en una solución. Una muestra sólida molida simplemente se presiona a la superficie del cristal ATR. Para muestras duras que pueden presentar un desafío para moler en un sólido fino, el área total en contacto con el cristal puede verse comprometida a menos que se utilicen pequeños cristales ATR con durabilidad excepcional (por ejemplo, diamante de 2 mm). La pérdida de contacto con el cristal resultaría en una disminución de la intensidad de la señal debido a que la onda evanescente puede no penetrar efectivamente en la muestra. La longitud de trayectoria inherentemente corta del ATR debido a la profundidad de penetración corta (0.5-5 µm) permite que las muestras sólidas modificadas en la superficie se caractericen fácilmente con ATR.
Las muestras en polvo suelen ser tediosas de preparar para el análisis con espectroscopía de transmisión porque normalmente requieren que se conviertan en un pellet de KBr y garantizar que la muestra en polvo se muele lo suficiente para reducir la dispersión. Sin embargo, las muestras en polvo no requieren preparación de muestras cuando se toman los espectros ATR. Esto es ventajoso en términos de tiempo y esfuerzo, pero también significa que la muestra se puede recuperar fácilmente después del análisis.
Líquidos
La ventaja de usar ATR para analizar muestras líquidas se hace evidente cuando se requieren longitudes de trayectoria efectivas cortas. La reproducibilidad espectral de las muestras líquidas es cierta siempre que toda la longitud del cristal esté en contacto con la muestra líquida, asegurando que la onda evanescente interactúe con la muestra en los puntos de reflexión, y el grosor de la muestra líquida exceda la profundidad de penetración. Una pequeña longitud de trayectoria puede ser necesaria para las soluciones acuosas con el fin de reducir la absorbancia de agua.
Preparación de Muestras
ATR-FTIR se ha utilizado en campos que abarcan el análisis forense hasta aplicaciones farmacéuticas e incluso la preservación del arte. Debido a su facilidad de uso y accesibilidad se puede utilizar ATR para determinar la pureza de un compuesto. Con solo una cantidad mínima de muestra, esta investigadora es capaz de recolectar un rápido análisis de su muestra y determinar si ha sido adecuadamente purificada o requiere un procesamiento posterior. Como se puede observar en la Figura\(\PageIndex{35}\), el tamaño de la muestra es minucioso y no requiere preparación. La muestra se coloca en estrecho contacto con el cristal ATR girando una perilla que aplicará presión a la muestra (Figura\(\PageIndex{36}\)).


ATR tiene una ventaja añadida en que encierra inherentemente la trayectoria óptica del haz IR. En un FTIR de transmisión, los compuestos atmosféricos están constantemente expuestos al haz IR y pueden presentar interferencia significativa con la medición de la muestra. Por supuesto, el FTIR de transmisión se puede purgar en un ambiente seco, pero la medición de la muestra puede volverse engorrosa. En una medición ATR, sin embargo, la luz del espectrómetro está constantemente en contacto con la muestra y la exposición al ambiente se reduce al mínimo.
Aplicación a Química Inorgánica
Una aplicación emocionante de ATR está en el estudio de las obras de arte clásicas. En el estudio de fragmentos de una obra de arte, donde las muestras son escasas y únicas en su tipo, el ATR es un método adecuado de caracterización porque solo requiere un pequeño tamaño de muestra. La determinación de los compuestos presentes en la técnica permite una conservación adecuada y una visión histórica de las piezas.
En un estudio que examinó varias muestras de pintura de diversos orígenes, se empleó un micro-ATR para el análisis. Este estudio utilizó un cristal de silicio con un índice de refracción de 2.4 y un tamaño de haz reducido. Más allá de un simple análisis de superficie, este estudio exploró la localización de diversos compuestos orgánicos e inorgánicos en las muestras mediante la realización de un análisis estratigráfico. Los investigadores lo hicieron incrustando las muestras tanto en KBr como en una resina de poliéster. Se compararon dos técnicas de incrustación para observar secciones transversales de las muestras. El mapeo de las muestras tomó aproximadamente 1-3 horas, lo que puede parecer bastante laborioso para algunos, pero considerando la naturaleza preciosa de la muestra, el tiempo de espera fue aceptable para los investigadores.
La imagen del microscopio óptico (Figura\(\PageIndex{37}\)) muestra una muestra de un área pintada de azul de la túnica de una estatua policromada italiana del siglo XIV de una Madonna. Los espectros mostrados en la Figura\(\PageIndex{38}\) se adquirieron de las diferentes capas que se muestran en la caja marcada en la Figura\(\PageIndex{37}\). Todos los espectros se recolectaron de la muestra en sección transversal y el mapa de color falso en cada espectro indica la ubicación de cada uno de estos compuestos dentro de la muestra incrustada. Los espectros corresponden a los compuestos inorgánicos enumerados en la Tabla\(\PageIndex{12}\), que también resalta bandas vibracionales características.

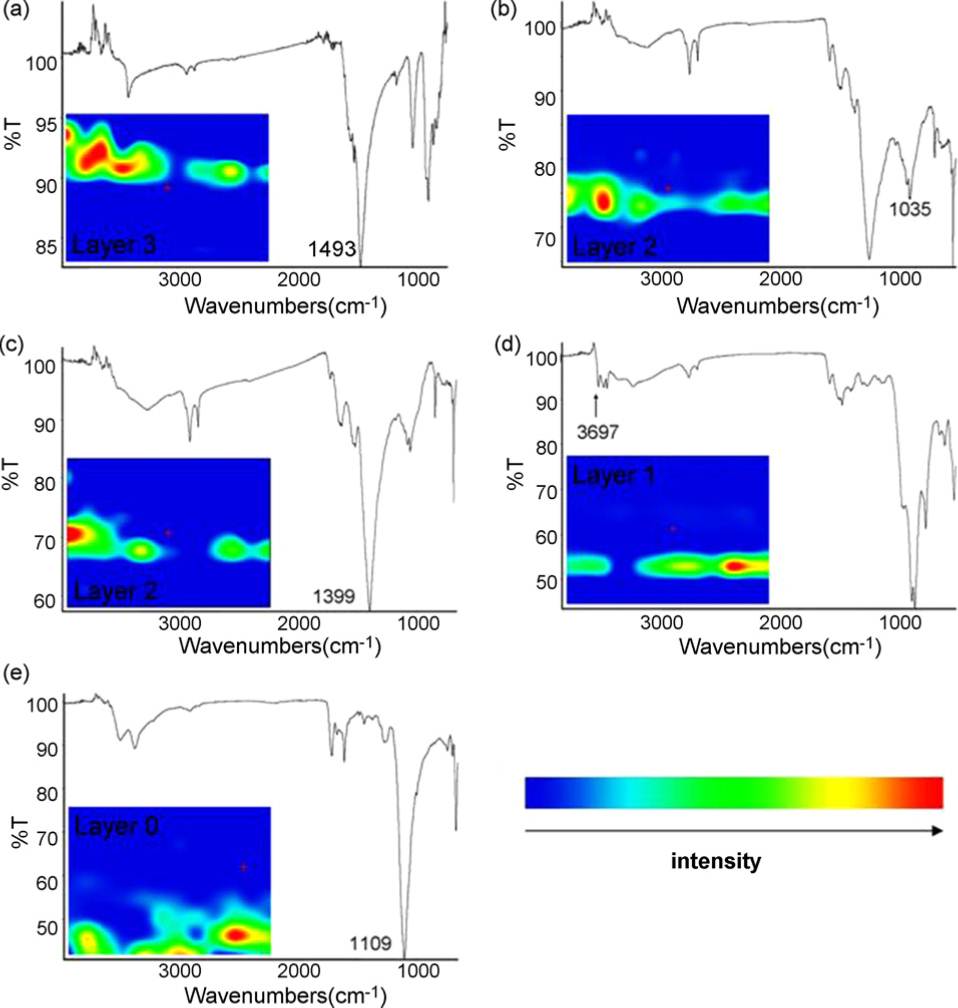
| Compuesto | Bandas espectrales seleccionadas | Asignación |
|---|---|---|
| Cu 3 (CO 3) 2 (OH) 2 (Azurita) | 1493 | CO 3 2 - Estiramiento asimétrico |
| Pigmentos azules a base de silicato | 1035 | Estiramiento Si-O |
| 2PbCo 3\(\cdot \) Pb (OH) 2 (Plomo blanco) | 1399 | CO 3 2 - Estiramiento asimétrico |
| Un pigmento rojo de silicato de aluminio ferruginoso natural (Bole) | 3697 | Estiramiento OH |
| CaSO 4\(\cdot \) (Yeso) | 1109 | SO 4 2 - estiramiento asimétrico |
La capa azul profundo 3 corresponde a azurita y la capa de pintura azul claro 2 a una mezcla de pigmentos azules a base de silicato y plomo blanco. Aunque más allá del límite de resolución espacial del cristal ATR de 20 µm, la absorción de bole fue detectada por las bandas de triple absorción características de 3697, 3651 y 3619 cm -1 como se ve en el espectro d de la Figura\(\PageIndex{37}\). La capa blanca 0 se identificó como yeso.
Para identificar el material aglutinante, la muestra incrustada en KBr demostró ser más efectiva que la resina de poliéster. Esto se debió en parte a la abrumadora absorbancia IR del yeso en el mismo rango espectral (1700-1600 cm -1) como estiramiento característico de la unión, así como a cierta absorción de contaminantes debido a la resina de incrustación de poliéster.
Para ubicar espacialmente pigmentos específicos y medios de unión, se realizó mapeo ATR en el área resaltada con una caja en la Figura\(\PageIndex{37}\). Las imágenes de color falso junto a cada espectro en la Figura\(\PageIndex{38}\) indican la presencia relativa del compuesto correspondiente a cada espectro en el área encajonada. El mapeo ATR se logró tomando 108 espectros en el área de 220x160 µm y seleccionando para cada compuesto identificado por su banda vibracional característica.