
4.2: Solvación Termodinámica
- Page ID
- 69671
Consideremos la termodinámica de un problema de solvatación acuosa. Esto ayudará a identificar diversos procesos físicos que ocurren en la solvatación, e identificar limitaciones a este enfoque. La solvatación se describe como el cambio en la energía libre para tomar el soluto de un estado de referencia, comúnmente tomado como el soluto aislado en vacío, en solución acuosa diluida:
Soluto (\(g\))\(\to\) Soluto (\(aq\))
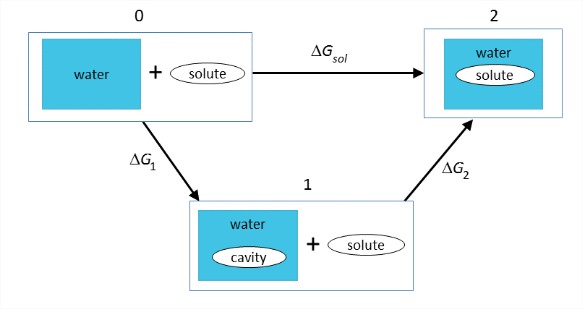
Conceptualmente, es útil dividir este proceso en dos etapas: (1) la energía requerida para abrir una cavidad en el líquido, y (2) la energía para poner el soluto en la cavidad y activar las interacciones entre soluto y disolvente.
Cada uno de estos términos tiene contribuciones entálpicas y entrópicas:
\[\begin{array} {rcl} {\Delta G_{\text{sol}}} & = & {\Delta H_{\text{sol}} - T \Delta S_{\text{sol}}} \\ {\Delta G_{\text{sol}}} & = & {\Delta G_1 + \Delta G_2} \\ {} & = & {\Delta H_1 - T\Delta S_1 + \Delta H_2 - T \Delta S_2} \end{array} \nonumber\]
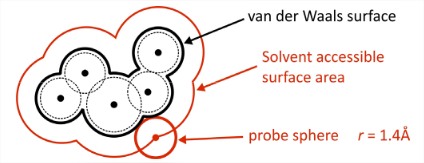
\(\Delta G_1\): Energía libre para abrir una cavidad en el agua. Estamos rompiendo las fuertes interacciones intermoleculares cohesivas en el agua (\(\Delta H_1\)), creando un vacío contra la presión constante y reduciendo la entropía configuracional de la red de enlaces de hidrógeno del agua (\(\Delta S_1\)). Por lo tanto\(\Delta G_1\) es grande y positivo.El efecto hidrofóbico está dominado por este término. En los modelos atomísticos, las cavidades para biomoléculas se definen comúnmente a través del área de superficie accesible al solvente del soluto (SASA). Para dar cuenta del volumen excluido en la escala de distancia de una molécula de agua, el SASA se puede obtener rodando una esfera con radio 1.4\(\mathring{A}\) sobre la superficie de van der Waals del soluto.
\(\Delta G_2\): Energía libre para insertar el soluto en la cavidad, activar las interacciones entre soluto y disolvente. La solvatación iónica y polar suele estar dominada por este término. Esto incluye las interacciones electrostáticas y H-bond favorables (\(\Delta H_2\)). También puede incluir una reestructuración del soluto y/o solvente en su interfaz debido a los nuevos cargos. El tratamiento más simple de este proceso describe el disolvente puramente como un medio dieléctrico homogéneo y el soluto como una simple esfera o cavidad incrustada con cargas puntuales o dipolos. Se originó a partir del ciclo Born-Haber utilizado por primera vez para describir\(\Delta H_{rxn}\) iones en fase gaseosa, y formó la base de numerosos enfoques de solvatación de continuidad y cavidad en continuo.
Dada la gran cantidad de efectos competidores que involucran interacciones soluto, solvente e intermoleculares, predecir el resultado de este proceso es complicado.
Mirar el ciclo anterior ilustra muchas de las complicaciones de este enfoque relevantes para la biofísica molecular, incluso sin preocuparse por los detalles atomísticos. Desde un punto de vista práctico, los dos pasos de este ciclo a menudo pueden tener gran magnitud pero signo opuesto, resultando en un alto nivel de incertidumbre sobre\(\Delta G_{\text{sol}}\) —incluso su signo! Más importante aún, este ciclo simplificado supone que se puede trazar un límite limpio entre el soluto y el disolvente, el área de superficie accesible al disolvente. También asume que la influencia del disolvente es perturbadora, en el sentido de que el disolvente no influye en la estructura del soluto o que existe trastorno conformacional o flexibilidad en el soluto y/o disolvente. Sin embargo, se pueden utilizar ciclos termodinámicos aún más detallados para abordar algunas de estas limitaciones:
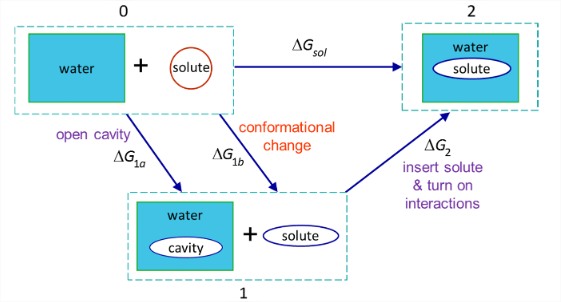
\(\Delta G_{1a}\): Energía libre para crear una cavidad en el agua para la molécula solvatada final.
\(\Delta G_{1b}\): Energía libre para inducir el cambio conformacional al soluto para el estado solvatado final.
\(\Delta G_{2}\): Energía libre para insertar el soluto en la cavidad, activar las interacciones entre soluto y disolvente. Esto incluye activar interacciones electrostáticas y enlaces de hidrógeno, así como permitir que el disolvente se reorganice alrededor del soluto:
\[\Delta G_{2} = \Delta G_{\text{solute-solvent}} + \Delta G_{\text{solvent reorg}}\nonumber\]
La entropía configuracional puede ser importante para cada paso de este ciclo, y se puede calcular usando 1
\[S=-k_{B} \sum_{i} P_{i} \ln P_{i} \nonumber\]
Aquí la suma es sobre las probabilidades de microestado, que se pueden expresar en términos de la probabilidad conjunta del soluto con una conformación dada y la probabilidad de una configuración de disolvente dada alrededor de esa estructura de soluto. En la etapa 1, se puede promediar sobre la entropía conformacional del disolvente para la forma de la cavidad (1a) y la conformación del soluto (1b). La etapa 2 incluye la variación configuracional del disolvente y la variación acompañante en la fuerza de interacción.
Con un conocimiento de termodinámica de solvatación para diferentes especies, es posible construir ciclos termodinámicos para una variedad de procesos de solvatación relacionados:
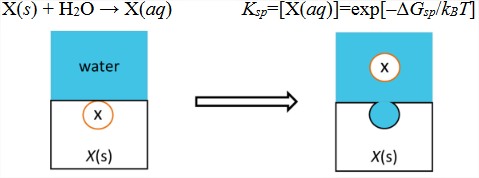
1) Solubilidad. El equilibrio entre la molécula en su forma sólida y en solución se cuantifica a través del producto de solubilidad\(K_{sp}\), que depende del cambio de energía libre de transferencia entre estas fases
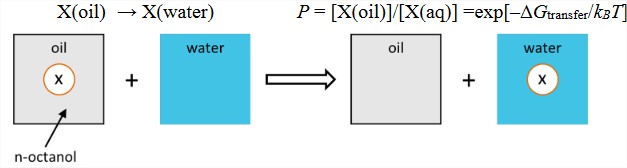
2) Transfiere energía libre. La forma empírica más común de cuantificar la hidrofobicidad es medir la partición de un soluto entre aceite y agua. El coeficiente de partición\(P\) está relacionado con la energía libre necesaria para transferir un soluto del disolvente no polar (típicamente octanol) al agua
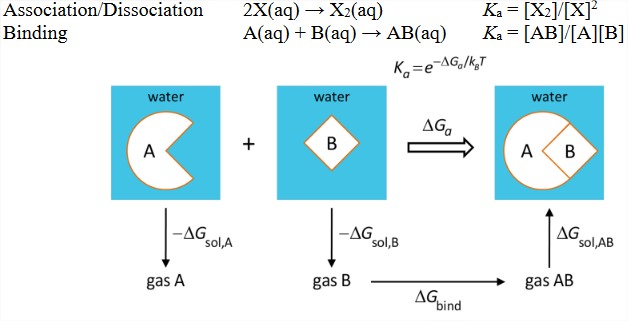
3) Procesos de asociación bimolecular
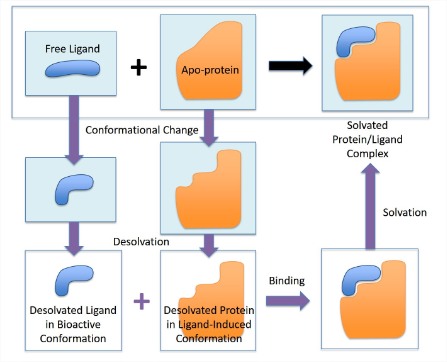
Encuadernación con selección conformacional
____________________________________________
- Ver C. N. Nguyen, T. K. Young y M. K. Gilson, Teoría de la solvatación no homogénea de Grid: Estructura de hidratación y termodinámica de la cucurbitácea del receptor miniatura [7] urilo, J. Chem. Phys. 137 (4), 044101 (2012).