
15.2: Transiciones Vibración-Rotación
- Page ID
- 74423

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Cuando los estados electrónicos inicial y final son idénticos, pero los respectivos estados vibracionales y rotacionales no lo son, se trata de transiciones entre estados de vibración-rotación de la molécula. Estas transiciones se estudian en espectroscopía infrarroja (IR) utilizando luz de energía en el\(^{-1} \text{ (far IR) to 5000 cm}^{-1}\) rango de 30 cm. El análisis del elemento de matriz dipolo eléctrico aún comienza con la integral del momento dipolo electrónico,\(\langle \psi_{ei} | \mu | \psi_{ei} \rangle = \mu (\textbf{R}),\) pero la integración sobre las coordenadas vibracionales internas ya no produce el momento dipolar promediado vibracionalmente. En cambio, se forma la integral del dipolo de transición vibracional:
\[ \langle \chi_{vf} | \mu (\textbf{R} ) | \chi_{vi} \rangle = \mu_{f,i} \]
entre los estados\( \chi_i \text{ and final } \chi_f \) vibracionales iniciales.
Los Derivados del Momento Dipolo
Expresando\(\mu ( \textbf{R} )\) en una expansión de serie de potencias sobre la posición de la longitud del enlace de equilibrio (denotada\(\textbf{R}_e\) colectiva e\(R_{a,e}\) individualmente):
\[ \mu ( \textbf{R} ) = \mu( \textbf{R}_e ) + \sum\limits_a \dfrac{\partial \mu}{\partial R_a}(R_a - R_{a,e}) + ..., \]
sustituyendo en la\( \langle\chi_{vf} | \mu ( \textbf{R} ) | \chi_{vi} \rangle \) integral, y utilizando el hecho de que\(\chi_i \text{ and } \chi_f\) son ortogonales (porque son funciones propias del movimiento vibratorio en la misma superficie electrónica y por lo tanto del mismo hamiltoniano vibracional), se obtiene:
\[ \langle \chi_{vf} | \mu ( \textbf{R} ) | \chi_{vi} \rangle = \mu ( \textbf{R}_e ) \langle \chi_{vf} | \chi_{vi} \rangle + \sum\limits_a \dfrac{\partial \mu}{\partial \textbf{R}_a} \langle \chi_{vf} | (R_a - R_{a,e}) | \chi_{vi} \rangle + ... \]
\[ = \sum\limits_a \left( \dfrac{\partial \mu}{\partial \textbf{R}_a}\right) \langle\chi_{vf} | (\textbf{R}_a - \textbf{R}_{a,e}) | \chi_{vi} \rangle + ... . \]
Este resultado puede interpretarse de la siguiente manera:
- Cada modo vibracional independiente de la molécula contribuye al\( \mu_{f,i} \) vector una cantidad igual a\( \left( \dfrac{\partial \mu}{\partial \textbf{R}_a} \right) \langle \chi_{vf} | (\textbf{R}_a - \textbf{R}_{a,e}) | \chi_{vi} \rangle + ... . \)
- Cada contribución de este tipo contiene una parte\( \left( \dfrac{\partial \mu}{\partial \textbf{R}_a} \right) \) que depende de cómo la función del momento dipolo de la molécula varía con la vibración a lo largo de ese modo particular (etiquetada a),
- y una segunda parte\( \langle\chi_{vf} | ( \textbf{R}_a - \textbf{R}_{a,e} ) | \chi_{vi} \rangle \) que depende del carácter de las funciones de onda vibracionales iniciales y finales.
Si la vibración no produce una modulación del momento dipolar (e.g., como ocurre con la vibración de estiramiento simétrico de la\(CO_2\) molécula), su intensidad infrarroja desaparece porque\( \left( \dfrac{\partial \mu}{\partial \textbf{R}_a} \right) = 0. \) Uno dice que tales transiciones son infrarrojas “inactivas”.
Reglas de selección sobre v en la aproximación armónica
Si las funciones vibracionales se describen dentro de la aproximación del oscilador armónico, se puede demostrar que las\( \langle\chi_{vf} | ( \textbf{R}_a - \textbf{R}_{a,e} ) | \chi_{vi}\rangle \) integrales desaparecen a menos que vf = vi +1, vi -1 (y que estas integrales son proporcionales a\(\sqrt{(vi +1)} \text{ and } \sqrt{(vi)}\) en los casos respectivos). Incluso cuando\(\chi_{vf} \text{ and } \chi_{vi}\) son bastante no armónicos, resulta que tales\(\Delta v = \pm 1\) transiciones tienen las\( \langle\chi_{vf} | (\textbf{R}_a - \textbf{R}_{a,e}) | \chi_{vi} \rangle \) integrales más grandes y por lo tanto las intensidades infrarrojas más altas. Por estas razones, las transiciones que corresponden a\(\Delta v = \pm 1\) se denominan "fundamentales “; a las que\(\Delta v = \pm 2\) se les llama transiciones de “primer armónico”.
En resumen entonces, las vibraciones para las cuales el momento dipolar de la molécula se modula a medida que ocurre la vibración (\(\left( \frac{\partial \mu}{\partial \textbf{R}_a}\right)\)es decir, para las cuales no es cero) y para las cuales\(\Delta v = \pm 1\) tienden a tener grandes intensidades infrarrojas; los armónicos de tales vibraciones tienden a tener intensidades menores, y aquellas para las cuales \( \left( \frac{\partial \mu}{\partial \textbf{R}_a} \right) = 0 \)no tienen intensidad.
Reglas de selección rotacional
El resultado de todas las contribuciones de los modos vibracionales a
\[ \sum\limits_a \left( \frac{\partial \mu}{\partial \textbf{R}_a} \right) \langle \chi_{vf} | (\textbf{R}_a - \textbf{R}_{a,e}) | \chi_{vi} \rangle \]
es un vector\(\mu_{trans}\) que se conoce como el momento vibratorio “dipolo de transición”. Este es un vector con componentes a lo largo, en principio, de los tres ejes internos de la molécula. Para cada transición vibracional particular (es decir, cada particular\(\chi_i \text{ and } \chi_f\)) su orientación en el espacio depende únicamente de la orientación de la molécula; por lo tanto, se dice que está bloqueada al marco de coordenadas de la molécula. Como tal, su orientación relativa a las coordenadas fijadas en laboratorio (que es necesaria para efectuar una derivación de reglas de selección rotacional como se hizo anteriormente en este Capítulo) puede describirse tanto como se hizo anteriormente para el momento dipolar promediado vibracionalmente que surge en transiciones puramente rotacionales. Sin embargo, existen importantes diferencias de detalle. En particular,
- Para una molécula lineal\(\mu_{trans}\) puede tener componentes ya sea a lo largo (por ejemplo, cuando se excitan vibraciones de estiramiento; estos casos se denotan\(\sigma\) -casos) o perpendiculares a (por ejemplo, cuando las vibraciones de flexión se excitan; se denotan\(\pi\) casos) el eje de la molécula.
- Para las especies superiores simétricas, no es\(\mu_{trans}\) necesario que se encuentre a lo largo del eje de simetría de la molécula; puede tener componentes a lo largo o perpendiculares a este eje.
- Para las cimas esféricas,\(\mu_{trans}\) desaparecerán siempre que la vibración no induzca un momento dipolar en la molécula. Las vibraciones como el movimiento de estiramiento\(a_1\) C-H totalmente simétrico en CH\(_4\) no inducen un momento dipolar y, por lo tanto, son inactivas por infrarrojos; las vibraciones no totalmente simétricas también pueden estar inactivas si no inducen ningún momento dipolar.
Como resultado de las consideraciones anteriores, las integrales angulares
\[ \langle \phi_{ir} | \mu_{trans} | \phi_{fr} \rangle = \int \text{D}_{\text{L, M, K}}(\theta ,\phi , \chi) \mu_{trans} \textbf{D}^{\text{*}}_{\text{L', M', K'}}(\theta , \phi , \chi)\text{ sin}\theta \text{ d}\theta \text{ d}\phi \text{ d}\chi \]
que determinan las reglas de selección rotacional apropiadas para las transiciones vibracionales producen resultados similares, pero no idénticos, como en el caso de transición puramente rotacional.
La derivación de estas reglas de selección procede como antes, con las siguientes consideraciones adicionales. Los\( \mu_{trans}\) componentes del momento dipolar de transición a lo largo de los ejes labfixed deben estar relacionados con sus coordenadas de moléculas fijas (que están determinadas por la naturaleza de la transición vibracional como se discutió anteriormente). Esta transformación, como se da en el texto de Zare, dice lo siguiente:
\[ (\mu_{trans})_m = \sum\limits_k \textbf{D}^{\text{*}}_{\text{l, m, k}}(\theta , \phi , \chi )(\mu_{trans})_k \]
donde\((\mu_{trans})_m \) con m = 1, 0, -1 se refieren a los componentes a lo largo de los ejes fijados en laboratorio (X, Y, Z) y\((\mu_{trans})_k\) con k = 1, 0, -1 se refieren a los componentes a lo largo de los ejes fijos de la molécula (a, b, c).
Esta relación, cuando se usa, por ejemplo, en la integral E1 superior simétrica o esférica:
\[ \langle \phi_{ir} | \mu_{trans} | \phi_{fr} \rangle = \int \text{D}_{\text{L,M,K}}(\theta , \phi , \chi )\mu_{trans} \textbf{D}^{\text{*}}_{\text{L', M', K'}}(\theta , \phi , \chi )\text{ sin}\theta \text{ d}\theta \text{ d}\phi \text{ d}\chi \]
da lugar a productos de 3-j símbolos de la forma:
\[ \left( \dfrac{\text{L' 1 L}}{\text{M' m -M}} \right) \left( \dfrac{\text{L' 1 L}}{\text{K' k -K}} \right). \]
El producto de estos símbolos 3-j no se desvanece solo bajo ciertas condiciones que proporcionan las reglas de selección rotacional aplicables a las líneas vibracionales de moléculas superiores simétricas y esféricas.
Ambos símbolos 3-j se desvanecerán a menos que
L = L' +1, L' o L'-1.
En el caso especial en el que L = L' =0 (y de ahí con M = M' =0 = K = K', lo que significa que m = 0 = k), estos símbolos 3-j vuelven a desaparecer. Por lo tanto, las transiciones con
L = L' =0
están nuevamente prohibidos. Como es habitual, el hecho de que el número cuántico fijo en laboratorio m pueda oscilar por encima de m = 1, 0, -1, requiere que
M = M' + 1, M', M'-1.
Las reglas de selección para\(\Delta \textbf{K}\) dependen de la naturaleza de la transición vibracional, en particular, del componente\(\mu_{trans}\) a lo largo de los ejes fijos de moléculas. Para que el segundo símbolo 3-j no se desvanezca, uno debe tener
K = K' + k,
donde k = 0, 1 y -1 se refieren a estos componentes fijados a moléculas del dipolo de transición. Dependiendo de la naturaleza de la transición, diversos valores k contribuyen.
Tops simétricos
En una molécula superior simétrica como\(NH_3\), si el dipolo de transición se encuentra a lo largo del eje de simetría de la molécula, solo k = 0 contribuye. Tales vibraciones preservan la simetría de la molécula con respecto a este eje de simetría (por ejemplo, el modo de estiramiento N-H totalmente simétrico en\(NH_3\)). Así se obtiene la regla\(\Delta K = 0\) de selección adicional. Además, para K = K' = 0, todas las transiciones con\(\Delta L = 0\) desaparecen porque el segundo símbolo 3-j desaparece. En resumen, uno tiene:
\( \Delta K = 0; \Delta M = \pm 1, 0; \Delta L = \pm 1,0\)(pero L = L' = 0 está prohibido y todos\(\Delta L = 0\) están prohibidos para K = K' = 0)
para cimas simétricas con vibraciones cuyo dipolo de transición se encuentra a lo largo del eje de simetría.
Si el dipolo de transición se encuentra perpendicular al eje de simetría, solo k = ±1 contribuye. En este caso, se encuentra
\( \Delta K = \pm 1; \Delta M = \pm 1, 0; \Delta L = \pm 1, 0 \)(ni L = L' = 0 ni K = K' = 0 pueden ocurrir para tales transiciones, por lo que no hay restricciones adicionales).
Moléculas Lineales
Cuando el análisis anterior se aplica a una especie diatómica como el HCl, solo k = 0 está presente ya que la única vibración presente en dicha molécula es la vibración de estiramiento de enlaces, que tiene\(\sigma\) simetría. Además, las funciones rotacionales son armónicos esféricos (los cuales pueden verse como\(\textbf{D}^{\text{*}}_{\text{L',M',K'}}(\theta , \phi ,\chi )\) funciones con K' = 0), por lo que los números cuánticos K y K' son idénticos cero. Como resultado, el producto de 3-j símbolos
\[ \left( \dfrac{\text{L' 1 L}}{\text{M' n -M}} \right) \left( \dfrac{\text{L' 1 L}}{K' k -K} \right) \]
reduce a
\[ \left( \dfrac{\text{L' 1 L}}{\text{M' m -M}} \right) \left( \dfrac{\text{L' 1 L}}{\text{0 0 0}} \right) \]
que se desvanecerá a menos que
\[ \text{ L = L' + 1, L' - 1, }\]
pero no L = L' (ya que la paridad hace que el segundo símbolo 3-j desaparezca), y
\[ \text{ M = M' + 1, M', M' - 1. } \]
Las transiciones L = L'+1 se denominan absorciones de ramificación R y las que obedecen a L = L' -1 se denominan transiciones de ramificación P. De ahí que las reglas de selección
\[ \Delta M = \pm 1, 0; \Delta L = \pm 1 \]
son idénticos a los de las transiciones puramente rotacionales.
Cuando se aplican a moléculas poliatómicas lineales, estas mismas reglas de selección resultan si la vibración es de\(\sigma\) simetría (es decir, tiene k = 0). Si, por otro lado, la transición es de\(\pi\) simetría (es decir, tiene k = ±1), entonces el dipolo de transición se encuentra perpendicular al eje de la molécula, se obtiene:
\[ \Delta M = \pm 1,0; \Delta L = \pm 1,0. \]
Estas reglas de selección se derivan al darse cuenta de que además de k = ±1, se tiene: (i) una función de onda rotacional de molécula lineal que en el nivel vibracional v = 0 se describe en términos de una matriz\(\text{D}_{\text{L',M',0}} (\theta , \phi ,\chi )\) de rotación sin momento angular a lo largo del eje molecular, K' = 0; (ii) una molécula v = 1 cuya la función de onda rotacional debe estar dada por una matriz de rotación\(\text{D}_{\text{L,M,1}} (\theta ,\phi , \chi )\) con una unidad de momento angular alrededor del eje de la molécula, K = 1. En este último caso, el momento angular es producido por la propia\(\pi\) vibración degenerada. Como resultado, las reglas de selección anteriores derivan del siguiente producto de 3-j símbolos:
\[ \left( \dfrac{\text{L' 1 L}}{\text{M' m -M}} \right) \left( \dfrac{\text{L' 1 L}}{\text{0 1 -1}} \right). \]
Debido a que las\(\Delta L = 0\) transiciones están permitidas para\(\pi \text{ vibrations, one says that } \pi\) las vibraciones poseen ramas Q- además de sus ramas R- y P- (con\(\Delta L = 1 \text{ and } -1,\) respectivamente).
En la figura que se muestra a continuación, se muestra el espectro de absorción vibracional\(\rightarrow\) v = 0 v = 1 (fundamental) del HCl. Aquí los picos a menor energía (a la derecha de la figura) pertenecen a transiciones de rama P y ocurren a energías dadas aproximadamente por:
\[ E = \hbar \omega_{\text{stretch}} + \left( \dfrac{h^2}{8\pi^2I} \right)((L - 1)L - L(L + 1)) = \hbar \omega_{\text{stretch}} -2\left( \dfrac{h^2}{8\pi^2 I} \right)L. \]
Las transiciones de ramificación R ocurren a energías más altas dadas aproximadamente por:
\[ E = \hbar \omega_{\text{stretch}} + \left( \dfrac{h^2}{8\pi^2I} \right)((L+1)(L+2) - L(L + 1)) = \hbar \omega_{\text{stretch}} + 2\left( \dfrac{h^2}{8\pi^2I} \right)(L + 1). \]
La absorción que “falta” de la figura de abajo que se encuentra ligeramente por debajo de 2900 cm-\(^{-1}\) es la transición de rama Q para la cual L = L'; está ausente porque las reglas de selección lo prohíben.
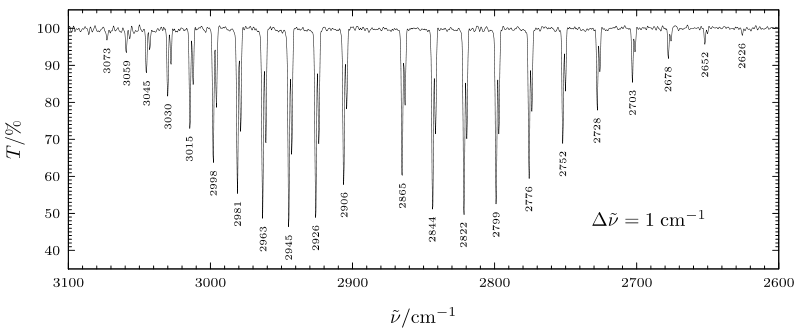
Cabe señalar que los espaciamientos entre los picos observados experimentalmente en HCl no son constantes como se esperaría con base en las fórmulas de ramificación P y R anteriores. Esto se debe a que el momento de inercia apropiado para el nivel vibracional v = 1 es diferente al del nivel v = 0. Estos efectos del acoplamiento vibración-rotación se pueden modelar permitiendo que los niveles v = 0 y v = 1 tengan energías rotacionales escritas como
\[ E = \hbar \omega_{\text{stretch}} \left( v + \dfrac{1}{2} \right) + \left( \dfrac{h^2}{8\pi^2I_v} \right) (L(L + 1)) \]
donde v y L son los números cuánticos vibracionales y rotacionales. Las energías de transición de las ramas P y R que pertenecen a estos niveles de energía pueden escribirse como:
\[ E_P = \hbar \omega_{\text{stretch}} - \left[ \left( \dfrac{h^2}{8\pi^2I_1} \right) + \left( \dfrac{h^2}{8\pi^2I_0}\right) \right] L + \left[ \left( \dfrac{h^2}{8\pi^2I_1} \right) - \left( \dfrac{h^2}{8\pi^2I_0} \right) \right] L^2 \]
\[ E_R = \hbar\omega_{\text{stretch}} + 2\left( \dfrac{h^2}{8\pi^2I_1} \right) + 3\left[ \left( \dfrac{h^2}{8\pi^2I_1}\right) - \left( \dfrac{h^2}{8\pi^2I_0} \right) \right]L + \left[ \left( \dfrac{h^2}{8\pi^2I_1}\right) - \left( \dfrac{h^2}{8\pi^2I_0}\right) \right]L^2. \]
Claramente, estas fórmulas se reducen a las mostradas anteriormente en el límite I1 = I0.
Si la longitud del enlace promediada vibracionalmente es más larga en el estado v = 1 que en el estado v = 0, que es de esperar,\(I_1\) será mayor que\(I_0\), y por lo tanto\( \left[ \left( \dfrac{h^2}{8\pi^2I_1} \right) - \left( \dfrac{h^2}{8\pi^2I_0} \right) \right] \) será negativa. En este caso, el espaciamiento entre las líneas de ramificación P vecinas aumentará como se muestra anteriormente para HCl. En contraste, el hecho de que\( \left[ \left( \dfrac{h^2}{8\pi^2I_1} \right) - \left( \dfrac{h^2}{8\pi^2I_0} \right) \right] \) sea negativo hace que el espaciamiento entre las líneas de ramificación R- vecinas disminuya, nuevamente como se muestra para HCl.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry and Jeff A. Nichols (Oak Ridge National Laboratory)