
16.4: La ley de Henry y la fugacidad y actividad de un componente de solución
- Page ID
- 74163

Al describir las actividades de los componentes de la solución, hemos tomado el estado estándar de componente\(A\) para ser el líquido puro a su presión de vapor de equilibrio\(P^{\textrm{⦁}}_A\),, a la temperatura de la solución. También podemos expresar la actividad utilizando la ley de Henry. La ley de Henry establece que la presión parcial de un componente por encima de su solución es directamente proporcional a la concentración del componente. Podemos elegir cualquier unidad conveniente para expresar la concentración de soluto. El valor de la constante de proporcionalidad depende de esta elección. Usando la fracción molar como unidad de concentración, la ley de Henry es
\[P_A=x_AP={\kappa }_Ay_A\]
(Ley de Henry)
donde la constante de proporcionalidad,\({\kappa }_A\), se llama la constante de la ley de Henry. (Cuando escribimos\(P_A=x_AP\), asumimos implícitamente que los componentes en fase gaseosa del sistema de equilibrio se comportan como gases ideales). La ley de Henry es más general que la ley de Raoult. En efecto, la ley de Raoult es un caso especial de la ley de Henry; si el soluto obedece la ley de Raoult, la constante de la ley de Henry lo es\(P^{\textrm{⦁}}_A\).
El valor de la constante de la ley de Henry depende de los componentes y de la temperatura. Experimentalmente, el valor de la constante se determina encontrando la pendiente de una parcela de\(P_A\) versus\(y_A\) en el límite as\(y_A\to 0\). El boceto de la Figura 5 ilustra la relación entre la\(P_A\) curva y la tangente de la ley de Henry a la misma en\(y_A=0\). Tenga en cuenta que la pendiente de la línea tangente at\(y_A=0\) es igual a su intersección en\(y_A=1\).
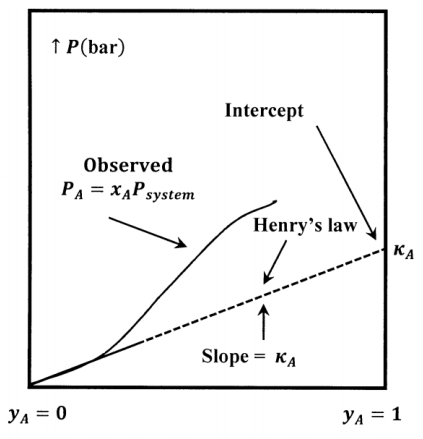
En la práctica, una concentración suficientemente baja de cualquier componente no electrolítico, en cualquier solución líquida, obedece a la ley de Henry. Por ello, nos referimos al componente que obedece a la ley de Henry como el soluto. Designamos el componente de mayor concentración como disolvente. La validez universal de la ley de Henry como aproximación de baja concentración tiene una interpretación física simple. La presión de vapor de soluto depende de los efectos netos de las fuerzas intermoleculares soluto-soluto, soluto-solvente y solvente-solvente.
Si todas estas fuerzas intermoleculares son iguales, las interacciones intermoleculares que determinan la composición en fase gaseosa son las mismas para las moléculas de disolvente que para las moléculas de soluto; las presiones de vapor del disolvente y el soluto son las mismas; el gas por encima de la solución tiene la misma composición que la solución; la presión parcial del soluto es proporcional a la concentración de soluto\(y_A\); la constante de proporcionalidad es la presión de vapor de disolvente puro,\(P^{\textrm{⦁}}_A\) y la solución obedece a la ley de Raoult. Sin embargo, si las fuerzas intermoleculares no son todas iguales, su efecto neto cambia a medida que cambia la concentración de soluto. A medida que aumenta la concentración de soluto, los efectos de las interacciones soluto-soluto se vuelven cada vez más importantes. Si estos son diferentes de los efectos de las interacciones soluto-disolvente y solvente-disolvente, la presión parcial de soluto no es proporcional a la concentración de soluto.
Por el contrario, a una concentración suficientemente baja, las moléculas de soluto están tan separadas que los efectos de las interacciones soluto-soluto se vuelven insignificantes. Solo las interacciones soluto—disolvente y solvente-disolvente afectan la presión de vapor del soluto. Debido a que estos permanecen constantes a medida que la concentración de soluto disminuye aún más, la presión parcial de soluto es proporcional a la concentración de soluto en este régimen de baja concentración. Sin embargo, si los efectos de las interacciones soluto-disolvente son diferentes de los de las interacciones solvente-disolvente, la presión de vapor de disolvente puro no\(P^{\textrm{⦁}}_A,\) es la constante de proporcionalidad.
Podemos suponer la existencia de un hipotético estado gaseoso ideal para cualquier sustancia, incluso una sustancia que no tenga presión de vapor medible a ninguna temperatura alcanzable. Podemos suponer que la sustancia tiene una energía de formación libre de Gibbs bien definida en este estado aunque no haya posibilidad de medir su valor. De igual manera, podemos suponer que cualquier soluto ejerce cierta presión parcial sobre su solución. Consideramos que esta presión parcial tiene algún valor finito, aunque sea demasiado pequeña para medirla. De ello se deduce que la fugacidad del soluto tiene un valor finito. La ley de Henry implica que la fugacidad es proporcional a la concentración de soluto, al menos en el límite de concentración arbitrariamente baja. Expresando la concentración de soluto A como su fracción molar, tenemos
\[f_A\left(P,y_A,y_B\right)={P_A}/{P^o}={x_AP}/{P^o}={{\kappa }_Ay_A}/{P^o}\]
y el potencial químico del soluto es
\[{\mu }_A\left(P,y_A,y_B\right)={\Delta }_fG^o\left(A,{HIG}^o\right)+RT{ \ln f_A\ }\left(P,y_A,y_B\right)\]
Si el soluto se comporta idealmente en fase gaseosa, la ley de Henry conduce a expresiones simples por su potencial y actividad química. El desarrollo de estas expresiones es muy similar al desarrollo correspondiente de la ley de Raoult. Las diferencias esenciales surgen de la introducción de un estado estándar diferente para la actividad del soluto. Comenzamos con nuestra ecuación básica para el potencial químico de\(\ A\) en fase gaseosa:\[{\mu }_A\left(g,P,x_A,x_B\right)={\Delta }_fG^o\left(A,{HIG}^o\right)+RT{ \ln \left[\frac{x_AP}{P^o}\right]\ }+\int^P_0{\left(\frac{{\overline{V}}_A\left(g\right)}{RT}-\frac{1}{P}\right)}dP\]
El término integral se desvanece porque asumimos el comportamiento ideal del gas. Escribimos\({\tilde{a}}_A\left(\mathrm{solute},P,y_A,y_B\right)\) para representar la actividad de\(A\) en una solución a presión\(P\) y en la que la composición se especifica por las fracciones molares\(y_A\) y\(y_B\).
Elegimos un líquido hipotético para que sea el estado estándar para la actividad del soluto. Este líquido hipotético es líquido puro\(A\) a la presión de vapor que exhibiría si siguiera la ley de Henry en toda la gama de posibles composiciones del sistema. Esta presión es igual a su constante de ley de Henry\({\kappa }_A\). (Ver Figura 5.) Denotemos el potencial químico de este estado estándar por\({\widetilde{\mu }}^o_A\left(Hyp\ \ell ,{\kappa }_A\right)\). El potencial químico del soluto\(A\) y la actividad de\(A\) en la solución están relacionados por
\[{\mu }_A\left(P,y_A,y_B\right)={\widetilde{\mu }}^o_A\left(Hyp\ \ell ,{\kappa }_A\right)+RT{ \ln {\tilde{a}}_A\ }\left(P,y_A,y_B\right)\]
y ya que\({\mu }_A\left(g,P,x_A,x_B\right)={\mu }_A\left(P,y_A,y_B\right)\), y el gas es ideal, tenemos
\[{ \ln \left[{\tilde{a}}_A\left(P,y_A,y_B\right)\right]\ }=\frac{{\Delta }_fG^o\left(A,{HIG}^o\right)-{\widetilde{\mu }}^o_A\left(Hyp\ \ell ,{\kappa }_A\right)}{RT}+{ \ln \left[\frac{x_AP}{P^o}\right]\ }\]
A excepción del estado estándar del soluto, esta es la misma que la ecuación que desarrollamos en la Sección 16.1.
Esta ecuación debe dar la actividad del soluto\(A\) en su estado estándar, que es puro líquido hipotético a una presión igual a la constante de la ley de Henry:\(P=P_A={\kappa }_A\). En este estado,\(x_A=y_A=1\) y\(x_B=y_B=0\). En su estado estándar, la actividad del soluto\(A\) es la unidad; tenemos\({\tilde{a}}_A\left(P,y_A,y_B\right)={\tilde{a}}_A\left({\kappa }_A,1,0\right)=1\). Haciendo estas sustituciones y reordenando, encontramos
\[\frac{{\Delta }_fG^o\left(A,{HIG}^o\right)-{\widetilde{\mu }}^o_A\left(Hyp\ \ell ,{\kappa }_A\right)}{RT}=-{ \ln \left[\frac{{\kappa }_A}{P^o}\right]\ }\]
Sustituyendo este resultado en nuestra ecuación general por\({ \ln \left[{\tilde{a}}_A\left(\mathrm{solute},P,y_A,y_B\right)\right]\ }\), encontramos que la actividad del soluto\(A\) es\[{\tilde{a}}_A\left(P,y_A,y_B\right)=\frac{x_AP}{{\kappa }_A}\] (cualquier solución, gas ideal)
El potencial químico del hipotético líquido puro de estado estándar cuya presión de vapor\(T\)\({\kappa }_A\) es
\[{\widetilde{\mu }}^o_A\left(Hyp\ \ell ,{\kappa }_A\right)={\Delta }_fG^o\left(A,{HIG}^o\right)+RT{ \ln \left[\frac{{\kappa }_A}{P^o}\right]\ }\]
Este es un resultado general para la actividad del soluto\(A\) cuando el estado estándar es el hipotético líquido puro cuya presión es\({\kappa }_A\), y se\(A\) comporta idealmente en la fase gaseosa. Si se obedece la ley de Henry, la tenemos\(x_AP=y_A{\kappa }_A\). Sustituyendo, encontramos
\[{\tilde{a}}_A\left(P,y_A,y_B\right)=y_A\](Se obedece la ley de Henry)
El desarrollo de la ley de Henry y el desarrollo de la ley de Raoult dan el mismo valor a la actividad química. Sin embargo, los estados estándar son diferentes.
Si el soluto obedece la ley de Raoult, el estado estándar que elegimos para el soluto es el soluto puro a su presión de vapor de equilibrio; en este estado, el soluto puro está en equilibrio con su propio gas. Las sustancias reales pueden satisfacer esta condición. Si el soluto obedece la ley de Henry, el estado estándar que elegimos para el soluto es un hipotético soluto de líquido puro a una presión igual a\({\kappa }_A\). En este estado, asumimos que el hipotético líquido está en equilibrio con su propio gas, también a presión\({\kappa }_A\). El hipotético líquido en estado estándar es una sustancia en la que las interacciones entre\(A\) moléculas tienen los mismos efectos que las interacciones, en una solución muy diluida, entre\(A\) las moléculas y las\(B\) moléculas que comprenden el disolvente. Si las soluciones de\(A\) y\(B\) son mal descritas por la ley de Raoult, la presión de vapor del líquido puro\(A\)\(P^{\textrm{⦁}}_A\),, es probable que sea muy diferente de la presión de vapor\({\kappa }_A\),, del hipotético líquido de estado estándar que definimos usando la ley de Henry.
Nuestro desarrollo produce un modelo para el potencial químico del soluto en el que la actividad es igual a la fracción molar de soluto. A la misma fracción molar, cada soluto tiene la misma actividad. Los potenciales químicos de diferentes solutos varían porque el potencial químico en su estado estándar es diferente para cada soluto. Concluimos que podemos permitir\({\tilde{a}}_A=y_A\) cualquier soluto suficientemente diluido, incluso cuando no es factible medir experimentalmente el potencial químico del soluto en su estado estándar.