
16.10: Propiedades coligativas - Elevación de punto de ebullición
- Page ID
- 74123

El sistema que imaginamos cuando hablamos de elevación del punto de ebullición se describe esquemáticamente en la Figura 7. Consideramos una solución de dos componentes,\(A\) y\(B\). Las fracciones molares de\(A\) y\(B\),\(y_A\) y\(y_B\), especifican la composición de la solución. Suponemos que uno de los componentes está presente a baja concentración. Llamamos a este componente el soluto, y lo designamos como compuesto\(A\). Bajo estos supuestos, tenemos\(y_A\approx 0\) y\(y_B=1-y_A\approx 1\). Suponemos además que\(A\) es no volátil, con lo que nos referimos a que la presión de vapor de puro\(A\)\(P^{\textrm{⦁}}_A\),, es muy pequeña. Entonces el segundo componente,\(B\), comprende la mayor parte del material del sistema. Llamamos componente\(B\) el solvente. Suponemos que la\(A\) —\(B\) solución está en equilibrio con una fase gaseosa. En principio, moléculas de ambos componentes están presentes en este gas. Ya que asumimos que esencialmente ningún componente\(A\) está presente en la fase gaseosa, tenemos\(x_A=0\) y\(x_B=1\). Suponemos también que la fase gaseosa\(B\) se comporta como un gas ideal y el soluto\(A\) obedece la ley de Henry.
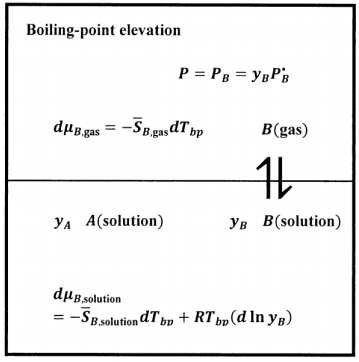
Cuando medimos el punto de ebullición de un sistema líquido, encontramos la temperatura a la que la presión de vapor del sistema se vuelve igual a un valor especificado. Para el punto de ebullición normal, esta presión es de 1 atmósfera, o 1.01325 bares. En el punto de ebullición, el disolvente en fase líquida está en equilibrio con el disolvente en fase gaseosa, de modo que el potencial químico del disolvente en fase líquida es igual al potencial químico del disolvente en fase gaseosa. Es decir, tenemos
\[\mu_{B,\mathrm{soluti}\mathrm{on}}=\mu_{B,\mathrm{gas}}.\nonumber \]
Queremos describir el cambio en la posición de equilibrio que se produce cuando hay un cambio incremental en la concentración de soluto\(dy_A\), mientras que la presión del sistema permanece constante. Si el sistema ha de mantenerse en equilibrio,\(\mu_{B,\mathrm{solution}}=\mu_{B,\mathrm{gas}}\) debe seguir siendo cierto. De ello se deduce que los potenciales químicos de las dos fases deben cambiar en tándem. El equilibrio continuado implica que\(d\mu_{B,\mathrm{solution}}=d\mu_{B,\mathrm{gas}}\) cuando la concentración de soluto cambia por\(dy_A\).
Podemos analizar el fenómeno de elevación del punto de ebullición para cualquier presión fija a la que el líquido puro\(B\) pueda estar en equilibrio con el gas puro\(B\). Designemos la presión fija como\(P^{\#}\). Designamos la temperatura del punto de ebullición del disolvente puro\(B\), en\(P^{\#}\), como\(T_B\); así\(P^{\#}=P^{\textrm{⦁}}_B\left(T_B\right)\), donde\(P^{\textrm{⦁}}_B\left(T_B\right)\) designa la presión de vapor de equilibrio del disolvente puro\(B\) a temperatura\(T_B\). Nuestro objetivo es encontrar la temperatura a la que una solución binaria está en equilibrio con el gas puro\(B\) a la presión fija\(P^{\#}\). Dejamos\(T_{bp}\) ser la temperatura de ebullición de la solución en\(P^{\#}\). La composición de la solución se especifica por la concentración de soluto,\(y_A=1-y_B\).
Dado que asumimos que el soluto obedece la ley de Henry, elegimos el estado estándar para que el\(A\) soluto sea el líquido hipotético puro\(A\) cuya presión de vapor está\({\textrm{ĸ}}_A\) en\(T\). Suponemos que eso\({\textrm{ĸ}}_A\) es sumamente pequeño. De la Sección 16. 4, entonces tenemos\(\tilde{a}_{A,\mathrm{solution}}=y_A\), para que
\[d ~ { \ln \tilde{a}_{A,\mathrm{solution}}\ }=d{ \ln y_A\ }\nonumber \]
a cualquier temperatura. De la Sección 16. 8, tenemos
\[d ~ { \ln \tilde{a}_{B,\mathrm{solution}}\ }=d{ \ln y_B\ }\nonumber \]
El disolvente puro en fase líquida está en equilibrio con el disolvente en fase gaseosa a\(P^{\#}=P^{\textrm{⦁}}_B\left(T_B\right)\) y\(T_B\). Imaginamos que creamos una solución añadiendo una pequeña cantidad de soluto\(A\), haciendo las concentraciones de soluto y disolvente\(y_A\) y\(y_B=1-y_A\), respectivamente. Mantenemos constante la presión del sistema\(P^{\#}\), mientras cambiamos la temperatura para mantener el equilibrio entre el disolvente en fase gaseosa y en fase de solución\(B\). La nueva temperatura es\(T_{bp}\).
La presión de la fase gaseosa B es constante a\(P^{\#}\). La temperatura va de\(T_B\) a\(T_{bp}\). Elegimos el estado estándar de actividad para que sea gas puro B a\(P^{\#}\) y T. Esto significa que la actividad del gas puro es unidad a cada temperatura, por lo que\(d{ \ln \tilde{a}_{B,\mathrm{gas}}\ }=0\).
Vale la pena señalar que podemos llegar a esta conclusión desde una perspectiva diferente: A partir de la Sección 14.14, el cambio incremental en la actividad es
\[d ~ { \ln \tilde{a}_{B,\mathrm{gas}}\ }=\left(-\frac{\overline{H}_B}{RT^2}+\frac{\tilde{H}^o_B}{RT^2}\right)dT\nonumber \]
donde\(\overline{H}_B\) es la entalpía molar parcial de la fase gaseosa\(B\) en\(T\), y\(\tilde{H}^o_B\) es la entalpía molar parcial de\(B\) en su estado estándar de actividad at\(\ T\). Ya que suponemos que la fase gaseosa es esencialmente pura\(B\), tenemos\(\overline{H}_B=\tilde{H}^o_B\) y, de nuevo,\(d \ln \tilde{a}_{B,\mathrm{gas}}=0\).
De la Sección 14.3, tenemos el resultado general de que
\[d\mu_B={\overline{V}}_BdP-{\overline{S}}_BdT+RT\left(d{ \ln \tilde{a}_B\ }\right)\nonumber \]
La presión y temperatura del sistema son\(P=P^{\#}\) y\(T=T_{bp}\). Tanto para la fase gaseosa como para la fase de solución, tenemos\(dP=0\) y\(dT=dT_{bp}\). Desde entonces\(d{ \ln \tilde{a}_{B,\mathrm{gas}}\ }=0\), tenemos
\[d\mu_{B,\mathrm{gas}}=-{\overline{S}}_{B,\mathrm{gas}}dT_{bp}\nonumber \]
Desde entonces\(d ~ { \ln \tilde{a}_{B,\mathrm{solution}}\ }=d{ \ln y_B\ }\), tenemos
\[d\mu_{B,\mathrm{solution}}=-{\overline{S}}_{B,\mathrm{solution}}dT_{bp}+RT_{bp}\left(d{ \ln y_B\ }\right)\nonumber \]
El potencial químico del disolvente puro, de presión constante, en fase gaseosa depende únicamente de la temperatura. El potencial químico del solvente en fase de solución a presión constante depende de la temperatura y concentración de soluto. El equilibrio se mantiene si
\[d\mu_{B,\mathrm{solution}}=d\mu_{B,\mathrm{gas}}\nonumber \]
Sustituyendo, tenemos
\[-{\overline{S}}_{B,\mathrm{solution}}dT_{bp}+RT_{bp}\left(d{ \ln y_B\ }\right)=-{\overline{S}}_{B,\mathrm{gas}}dT_{bp}\nonumber \]
Dado que\(y_B=1-y_A\approx 1\),
\[d{ \ln y_B\ }=d{ \ln \left(1-y_A\right)\ }={-dy_A}/{\left(1-y_A\right)}\approx -dy_A\nonumber \]
La relación\(d\mu_{B,\mathrm{solution}}=d\mu_{B,\mathrm{gas}}\) se convierte
\[-{\overline{S}}_{B,\mathrm{solution}}dT_{bp}-RT_{bp}dy_A=-{\overline{S}}_{B,\mathrm{gas}}dT_{bp}\nonumber \]
o
\[dy_A=\left(\frac{\overline{S}_{B,\mathrm{gas}}}-\overline{S}_{B,\mathrm{solution}} {RT_{bp}} \right)dT_{bp} \nonumber \]
Consideramos sistemas en los que el punto de ebullición de la solución\(T_{bp}\),, es poco diferente del punto de ebullición del solvente puro,\(T_B\). Entonces,\(T_B\approx T_{bp}\), y\({T_{bp}}/{T_B}\approx 1\). Dejamos\(\Delta T=T_{bp}-T_B\), dónde\(\left|\Delta T\right|\ll T_B\). Dado que la solución es casi pura\(B\), la entropía molar parcial de\(B\) en la solución es aproximadamente la de pura\(B.\) En consecuencia, esta diferencia de entropía molar parcial es, a una buena aproximación, solo la entropía de vaporización del disolvente, en equilibrio, en el punto de ebullición para la presión del sistema especificada,\(P^{\#}=P^{\textrm{⦁}}_B\left(T_B\right)\). Entonces, dado que la vaporización de puro\(B\) a\(P^{\#}\) y\(T_B\) es un proceso reversible,
\[{\left({\overline{S}}_{B,\mathrm{gas}}-{\overline{S}}_{B,\mathrm{solution}}\right)}_{P^{\#},T_{bp}}\approx {\left({\overline{S}}^{\textrm{⦁}}_{B,\mathrm{gas}}-{\overline{S}}^{\textrm{⦁}}_{B,\mathrm{liquid}}\right)}_{P^{\#},T_B}=\Delta_{\mathrm{vap}}S_B={\Delta_{\mathrm{vap}}H_B}/{T_B}\nonumber \]
para que
\[dy_A=\left(\frac{\Delta_{\mathrm{vap}}H_B}{RT_{bp}T_B}\right)dT_{bp}\nonumber \]
En la solución, la fracción molar de soluto es\(y_A\); en el disolvente puro, es cero. En\(P^{\#}\) y\(T_B\),\(\Delta_{\mathrm{vap}}H_B\) es una constante. Integrando, entre los límites\(\left(0,T_B\right)\) y\(\left(y_A,T_{bp}\right)\), tenemos
\[\int^{y_A}_0{dy_A}=\frac{\Delta_{\mathrm{vap}}H_B}{RT_B}\int^{T_{bp}}_{T_B}{\frac{dT_{bp}}{T_{bp}}}\nonumber \]y\[y_A=\frac{\Delta_{\mathrm{vap}}H_B}{RT_B}{ \ln \frac{T_{bp}}{T_B}\ }\nonumber \]
Presentando la aproximación\({ \ln x\approx x-1\ }\), que es válida para\(x\approx 1\), tenemos
\[y_A=\frac{\Delta_{\mathrm{vap}}H_B}{RT_B}\left(\frac{T_{bp}}{T_B}-1\right)=\frac{\Delta_{\mathrm{vap}}H_B}{RT^2_B}\Delta T\]
Resolviendo para\(\Delta T\),
\[\Delta T=\left(\frac{RT^2_B}{\Delta_{\mathrm{vap}}H_B}\right)y_A\nonumber \]
Dado que la entalpía de vaporización y la fracción molar son ambas mayores que cero,\(\Delta T=T_{bp}-T_B>0\); es decir, la adición de un soluto no volátil aumenta el punto de ebullición de un sistema líquido. Al medir\(\Delta T\), podemos encontrar\(y_A\); si conocemos la masa molar del disolvente, podemos calcular el número de moles de soluto en la solución. Si conocemos la masa del soluto utilizado para preparar la solución, podemos calcular la masa molar del soluto.
Frecuentemente es útil expresar la concentración de soluto como una molalidad en lugar de una fracción molar. Utilizando la relación dilución-solución entre la fracción molar y la molalidad de la Sección 16. 6,\(y_A={\overline{M}_B{\underline{m}}_A}/{1000}\), la elevación del punto de ebullición se convierte en:
\[\Delta T=\left(\frac{RT^2_B}{\Delta_{\mathrm{vap}}H_B}\right)\left(\frac{\overline{M}_B}{1000}\right){\underline{m}}_A\nonumber \]
Nuestra teoría predice que la elevación del punto de ebullición observada para un disolvente dado es proporcional a la concentración de soluto e independiente de las características moleculares del soluto. Los experimentos validan esta predicción; sin embargo, su precisión disminuye a medida que aumenta la concentración de soluto. Dejar
\[{\textrm{ĸ}}_B=\frac{RT^2_B}{\Delta_{\mathrm{vap}}H_B}\nonumber \]y\[{\textrm{ĸ}}^*_B=\frac{RT^2_B\overline{M}_B}{1000\ \Delta_{\mathrm{vap}}H_B}\nonumber \]
tenemos\(\Delta T={\textrm{ĸ}}_By_A\) y\(\Delta T={\textrm{ĸ}}^*_B{\underline{m}}_A\). Llamamos\({\textrm{ĸ}}_B\) o\({\textrm{ĸ}}^*_B\) la constante de elevación del punto de ebullición (o temperatura de ebullición) para el solvente\(B\). Para la determinación práctica de pesos moleculares, generalmente encontramos\({\textrm{ĸ}}_B\) o\({\textrm{ĸ}}^*_B\) midiendo el aumento en el punto de ebullición de una solución de composición conocida.