
8.4: Orbitales Moleculares para Diatómica Homonuclear
- Page ID
- 69431
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Si bien las formas específicas de los orbitales moleculares (su dependencia de r y z en un sistema de coordenadas cilíndricas) son diferentes para cada molécula, su dependencia del ángulo f como denota el número cuántico l y su g o u comportamiento con respecto a la inversión están completamente determinados por la simetría del sistema. Estas propiedades son comunes a todos los orbitales moleculares para moléculas diatómicas homonucleares. Además, el orden relativo de las energías orbitales es el mismo para casi todas las moléculas diatómicas homonucleares. Así podemos construir un diagrama de nivel de energía orbital molecular, similar al utilizado para construir las configuraciones electrónicas de los átomos en la tabla periódica. El diagrama de nivel de energía orbital molecular (Figura\(\PageIndex{1}\)) es tan fundamental para la comprensión de la estructura electrónica de las moléculas diatómicas como el diagrama orbital atómico correspondiente es para la comprensión de los átomos.
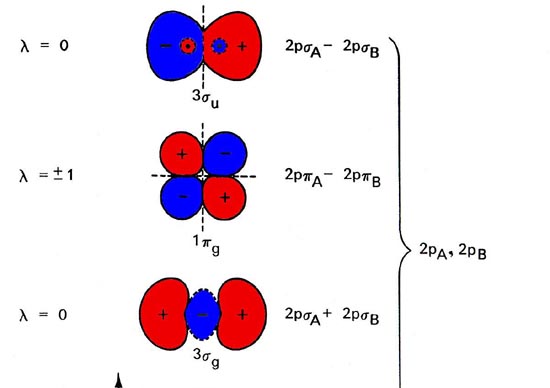


Figura\(\PageIndex{1}\): Diagrama de nivel de energía orbital molecular para moléculas diatómicas homonucleares que muestra la correlación de los orbitales moleculares con los orbitales atómicos de los átomos separados. La representación esquemática de los orbitales moleculares es para ilustrar sus formas generales y propiedades nodales (los nodos se indican con líneas discontinuas). Solo se muestra un componente de los orbitales degenerados 1pu y 1pg. El segundo componente es idéntico en forma en cada caso pero girado 90° fuera del plano. El orden de los niveles de energía orbitales mostrados en la figura se mantiene generalmente para todas las moléculas diatómicas homonucleares con la excepción de los niveles para los orbitales 1pu y 3sg, cuyo orden relativo se invierte para las moléculas posteriores\(C_2\).
Los orbitales moleculares exhiben las mismas propiedades generales que los orbitales atómicos, incluyendo una estructura nodal. Las propiedades nodales de los orbitales se indican en la Figura\(\PageIndex{1}\). Observe que las propiedades nodales reflejan correctamente el carácter g y u de los orbitales. La inversión de una g orbital intercambia regiones de signo similar y la órbita se deja sin cambios. La inversión de un orbital u intercambia las regiones positivas con las regiones negativas y la órbita cambia en signo.
Un orbital de una simetría particular puede aparecer más de una vez. Cuando esto ocurre, se agrega un número como prefijo al símbolo. Así hay 1 s g, 2 s g, 3 s g, etc. orbitales moleculares así como hay 1 s, 2 s, 3 s, etc. orbitales atómicos. El prefijo numérico es similar al número cuántico principal n en el caso atómico. A medida que n aumenta a través de un conjunto de simetría dado, por ejemplo, 1 s g, 2 s g, 3 s g, la energía orbital aumenta, la orbital aumenta de tamaño y consecuentemente se concentra densidad de carga más lejos de los núcleos, y finalmente el número de nodos aumenta a medida que n aumenta. Todas estas propiedades son comunes a los orbitales atómicos también.
Podemos obtener una comprensión cualitativa del diagrama de nivel de energía orbital molecular considerando el comportamiento de los orbitales bajo ciertas condiciones limitantes. El orbital molecular debe describir el movimiento del electrón para todos los valores de la separación internuclear; desde R = ¥ para los átomos separados, pasando por R = R e, el estado de equilibrio de la molécula, hasta R = 0, el átomo unido obtenido cuando los dos núcleos de la molécula se fusionan (en una reacción hipotética) para dar un solo núcleo. De ahí que un orbital molecular deba sufrir un cambio continuo de forma. Al límite de R grande debe reducirse a alguna combinación de orbitales atómicos dando la descripción orbital adecuada de los átomos separados y para R = 0 debe reducirse a un solo orbital atómico en el núcleo unido.
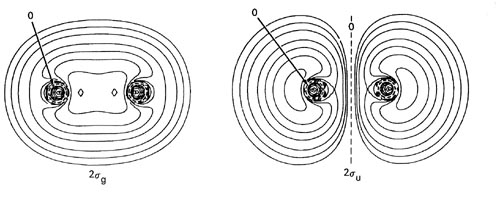
Consideremos, por ejemplo, el comportamiento limitante del orbital 1 s g en el caso de la molécula de hidrógeno. El estado más estable de H 2 se obtiene cuando ambos electrones se colocan en este orbital con espines emparejados dando la configuración electrónica 1 s g 2. Para grandes valores de la separación internuclear, la molécula de hidrógeno se disocia en dos átomos de hidrógeno. Así, la forma limitante del orbital molecular 1 s g para una separación infinita entre los núcleos debe ser una suma de orbitales de 1 s, uno centrado en cada uno de los núcleos. Si etiquetamos los dos núcleos como A y B podemos expresar la forma limitante del\(1s_g\) orbital como
\[1\sigma_g \rightarrow (1s_A + 1s_b\]
donde l s A es un orbital de 1 s centrado en el núcleo A, y l s B es un orbital l s centrado en el núcleo B. Esta forma para el orbital de 1 s g predice la distribución de densidad correcta para el sistema en grandes valores de R. Al cuadrar la función (1 s A + 1 s B) obtenemos para la densidad
\[ 1s_A 1s_A + 1s_B 1s_B +2 \times 1s_A 1s_B\]
Los dos primeros términos denotan que un electrón está en el átomo A y otro en el átomo B, ambos con distribuciones de densidad atómica de 1s. El término cruzado 2 'l s A' l s B obtenido en el producto es cero ya que la distancia entre los dos núcleos es tan grande que el solapamiento de los orbitales desaparece. Observe también que la función (l s A + l s B) tiene las mismas propiedades de simetría que el orbital molecular de 1 s g; es simétrica tanto con respecto a una rotación alrededor de la línea que une los núcleos como a una inversión de las coordenadas en el punto medio entre los núcleos. Se dice que el orbital de 1 s g para la molécula se correlaciona con la suma de orbitales de 1 s, uno en cada núcleo, para el caso de átomos separados.
Consideremos a continuación el caso limitante de los átomos separados para la molécula de helio. De los cuatro electrones presentes en He 2, dos se colocan en el orbital 1 s g y los dos restantes deben, por el principio de exclusión Pauli, colocarse en el siguiente orbital vacante de menor energía, el orbital 1 s u . La configuración electrónica de He 2 es así 1 s g 2 1 s u 2. El orbital de 1 s g se correlacionará con la suma de los orbitales de 1 s para los átomos de helio separados. De los dos electrones en el orbital molecular 1 s g uno se correlacionará con el orbital de 1 s en el átomo A y el otro con el orbital de 1 s sobre el átomo B. Dado que cada átomo de helio posee dos electrones de 1 s, el 1 s u orbital también debe correlacionar sus electrones con las funciones atómicas de 1 s en A y B. Además, la función correlacionada en este caso debe ser de simetría u. Una función con estas propiedades es
\[1\sigma_g \rightarrow (1s_A - 1s_b\]
La distribución de densidad limitante obtenida al cuadrar esta función coloca un electrón en una distribución atómica de 1 s en A, el otro en una distribución atómica de 1 s en B. La suma de las densidades de carga limitantes para los 1 s g y 1 s u orbitales moleculares colocan dos electrones en distribuciones de carga atómica de 1 s en cada átomo, la descripción adecuada de dos átomos de helio aislados.
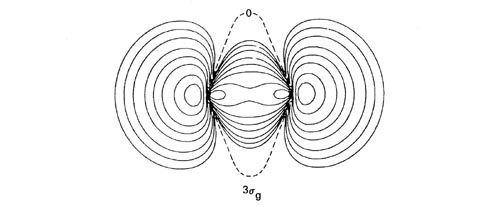
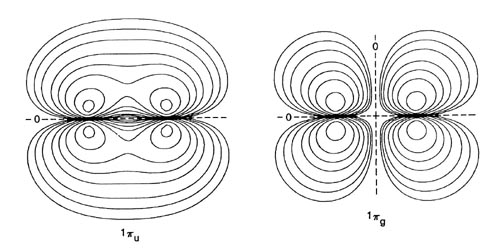
Cada orbital molecular homonuclear diatómico puede correlacionarse con la suma (para orbitales s g y p u) o la diferencia (para s u y p g orbitales) de orbitales similares en ambos átomos separados. Al llevar a cabo este procedimiento de correlación para cada orbital podemos construir un diagrama de correlación orbital molecular (Figura\(\PageIndex{2}\)) que relaciona cada uno de los niveles de energía orbitales en la molécula con los niveles de energía correlacionados en los átomos separados. Es importante señalar que la simetría de cada orbital se conserva en la construcción de este diagrama. Consideremos, por ejemplo, los orbitales moleculares que se correlacionan con los orbitales atómicos de 2 p. La dirección de aproximación de los dos átomos define un nuevo eje de cuantificación para los orbitales atómicos. El orbital 2 p que se encuentra a lo largo de este eje es de simetría s mientras que los dos orbitales restantes forman un conjunto degenerado de simetría p con respecto a este eje. La suma y diferencia de los orbitales 2 p s en cada centro se correlacionan con los orbitales 3 s g y 3 s u respectivamente, mientras que la suma y la diferencia de los orbitales 2 p p correlacionar con los orbitales p u y p g (Figura\(\PageIndex{2}\)).


Figura\(\PageIndex{2}\): Las formas atómicas separadas correlacionadas de los orbitales moleculares 3 s g, 3 s u , 1 p u y 1 p g. Los planos nodales se indican mediante líneas discontinuas. Sólo se muestra un componente de cada orbital p.
Para grandes valores de la distancia internuclear, cada orbital molecular se representa así por una suma o una diferencia de orbitales atómicos centrados en los dos átomos que interactúan. A medida que los átomos se acercan entre sí, los orbitales de cada átomo se distorsionan por los efectos de polarización y superposición. En general, las formas correlacionadas limitantes de los orbitales moleculares no son descripciones adecuadas de los orbitales moleculares para separaciones internucleares finitas.
Ahora estamos en condiciones de construir y determinar las configuraciones electrónicas de las moléculas diatómicas homonucleares agregando electrones dos a la vez a los orbitales moleculares con los espines de los electrones emparejados, llenando siempre primero los orbitales de menor energía. Al mismo tiempo, discutiremos la efectividad de cada orbital en la unión de los núcleos y haremos predicciones cualitativas respecto a la estabilidad de cada configuración molecular.
Hidrógeno. Los dos electrones en la molécula de hidrógeno pueden acomodarse ambos en el orbital de 1 s g si sus espines están emparejados y la configuración orbital molecular para H2 es de 1 s g 2 . Dado que el orbital 1 s g es el único orbital ocupado en el estado fundamental de H 2, la distribución de densidad mostrada anteriormente en la Figura\(\PageIndex{2}\) para H 2 es también la distribución de densidad para el 1 s g orbital cuando está ocupado por dos electrones. Los comentarios realizados previamente con respecto a la unión de los núcleos en H 2 por la distribución de carga molecular se aplican directamente a las propiedades de la densidad de carga de 1 s g. Debido a que concentra la carga en la región de unión y ejerce una fuerza de atracción sobre los núcleos, el orbital de 1 s g se clasifica como orbital de unión.
Las configuraciones electrónicas excitadas para moléculas pueden describirse y predecirse con la misma facilidad dentro del marco de la teoría orbital molecular que las configuraciones excitadas de átomos en la teoría orbital atómica correspondiente. Por ejemplo, un electrón en H2 puede ser excitado a cualquiera de los orbitales vacantes de mayor energía indicados en el diagrama de niveles de energía. La molécula excitada puede regresar a su configuración de tierra con la emisión de un fotón. La energía del fotón vendrá dada aproximadamente por la diferencia en las energías del orbital excitado y el orbital de estado fundamental de 1 s g. Así, tanto las moléculas como los átomos exhibirán un espectro lineal. El espectro lineal electrónico obtenido de una molécula se complica, sin embargo, por la aparición de muchas bandas laterales acompañantes. Estos tienen su origen en cambios en la energía vibratoria de la molécula que acompañan al cambio en la energía electrónica.
Helio. La configuración electrónica de He 2 es 1 s g 2 1 s u 2. Un orbital s u, a diferencia de un orbital s g, posee un nodo en el plano a medio camino entre los núcleos y perpendicular al eje de enlace. El orbital 1 s u y todos los orbitales s u en general, debido a esta propiedad nodal, no pueden concentrar la densidad de carga en la región de unión. En cambio, se concentra en la región antiunión detrás de cada núcleo (Figura\(\PageIndex{3}\)).

Figura\(\PageIndex{3}\): Mapas de contorno de las densidades de carga orbitales doblemente ocupadas\(1\sigma _g\) y\(1\sigma _u\) moleculares y de la distribución total de la carga molecular de\(He_2\) a R = 2.0 au. También se muestra un perfil de la distribución de carga total a lo largo del eje internuclear. Haga clic aquí para ver los valores de contorno.
Por lo tanto, los orbitales s u se clasifican como antiadherentes. Es evidente a partir de la forma de distribución de densidad para el orbital de 1 s u que la densidad de carga en este orbital separa los núcleos en lugar de unirlos. Generalmente, la ocupación de un número igual de orbitales s g y s u resulta en una molécula inestable. La fuerza de atracción ejercida sobre los núcleos por la densidad de carga en los orbitales s g no es suficiente para equilibrar tanto la fuerza nuclear de repulsión como la fuerza antiunión ejercida por la densidad en los orbitales s u. Así, la teoría orbital molecular atribuye la inestabilidad de He 2 a la ocupación igual de orbitales ligantes y antiadherentes. Observe que el principio de exclusión Pauli sigue siendo la causa básica de la inestabilidad. Si no fuera por el principio Pauli, los cuatro electrones podrían ocupar un orbital tipo s g y concentrar su densidad de carga en la región de baja energía potencial entre los núcleos. Es el principio Pauli, y no una cuestión de energética, lo que obliga a la ocupación del orbital antiadhesión 1 s u.
La distribución total de la carga molecular se obtiene sumando las densidades orbitales moleculares individuales para números de ocupación simple o doble según lo determinado por la configuración electrónica de la molécula. Así, la distribución de carga total para He 2 (Figura\(\PageIndex{3}\)) viene dada por la suma de las densidades orbitales de 1 s g y 1 s u para la doble ocupación de ambos orbitales. El efecto adverso que la propiedad nodal de la órbita 1 s u tiene sobre la estabilidad de He 2 es muy evidente en la distribución total de la carga. Se acumula muy poca densidad de carga en la porción central de la región de unión. El valor de la densidad de carga en el punto medio del enlace en He 2 es de sólo 0.164 au comparado con un valor de 0.268 au para H 2.
Debemos reconsiderar a la luz de la teoría orbital molecular la estabilidad de He 2 + y la inestabilidad de la molécula de hidrógeno con espines paralelos, casos discutidos previamente en términos de teoría de enlaces de valencia. El 2 + tendrá la configuración 1 s g 2 1 s u 1. Dado que el orbital de 1 s u solo está ocupado individualmente en He 2 +, se acumula menos densidad de carga en las regiones antiunión que la acumulada en estas mismas regiones en la molécula neutra. Así predominan las fuerzas de unión de la densidad de 1 s g doblemente ocupada y He 2 es estable. La configuración electrónica de H 2 es 1 s g 1 () 1 s u 1 () cuando los giros electrónicos son paralelos . Los electrones deben ocupar orbitales separados por el principio de exclusión de Pauli. Con igual ocupación de orbitales ligantes y antiadherentes, se predice que la especie H 2 () sea inestable.
Litio. La molécula Li 2 con la configuración 1 s g 2 1 s u 2 2 s g 2 marcas el inicio de lo que se puede llamar el segundo caparazón cuántico en analogía con el caso atómico. Dado que el orbital antiadhesión de 1 s u cancela aproximadamente la unión obtenida del orbital de unión de 1 s g, el enlace en Li 2 puede describirse como derivado del par único de electrones en el 2 s g orbital. La teoría de enlaces de valencia, o un modelo de Lewis para Li 2, también describe la unión en Li 2 como resultado de un enlace de par de electrones simples. Este es un resultado general. El número de enlaces predichos en una estructura simple de Lewis se encuentra a menudo para igualar la diferencia entre el número de orbitales de enlace ocupado y antiunión de la teoría orbital molecular.
Las formas de las distribuciones de densidad orbital para Li 2 (Figura\(\PageIndex{4}\)) confirman la predicción de que un enlace de par de electrones único es responsable de la unión en esta molécula.
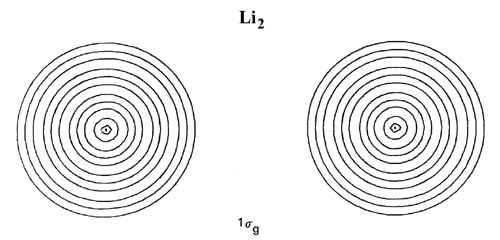
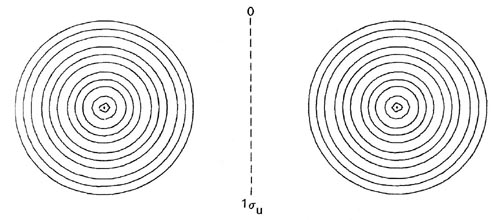
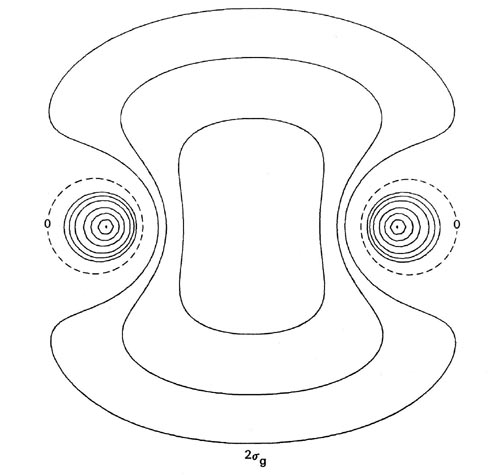
Figura\(\PageIndex{4}\): Mapas de contorno de las densidades de carga orbitales moleculares de 1 s g, 1 s u y 2 s g doblemente ocupadas para Li 2 a R = 5.051 au, la separación internuclear en equilibrio. Haga clic aquí para ver los valores de contorno. La distribución de carga molecular total para Li 2 se muestra en la Fig. 7-3.
Las distribuciones de densidad de 1 s g y 1 s u están fuertemente localizadas en las regiones de los núcleos con contornos esféricos característicos de distribuciones atómicas de 1 s. La adición de solo las densidades orbitales de 1 s g y 1 s u doblemente ocupadas en Li 2 dará una distribución que se asemeja muy estrechamente y puede identificarse con los 1 s doblemente ocupados o densidades atómicas de la cubierta interna en cada núcleo de litio. Solo la densidad de carga del par de electrones de valencia en el orbital de 2 s g se deslocaliza sobre la totalidad de la molécula y se acumula en cualquier medida en la región de unión.
Así, hay casos en los que los orbitales moleculares incluso en la longitud del enlace de equilibrio se asemejan estrechamente a sus formas atómicas limitantes Esto ocurre para los orbitales moleculares de la capa interna que se correlacionan con los orbitales atómicos de la capa interna en los átomos separados. Los electrones de la cubierta interna 1 s están unidos muy estrechamente al núcleo, ya que experimentan casi la carga nuclear completa y los radios efectivos de las distribuciones de densidad de 1 s son menores que las longitudes de enlace molecular. Debido a su estrecha unión y extensión restringida en el espacio, los electrones internos no participan en gran medida en la unión de una molécula. Así, con la excepción de H 2 y He 2 y sus iones moleculares, los orbitales moleculares 1 s g y 1 s u degeneran en orbitales de tipo atómico no superpuestos centrados en los dos núcleos y están clasificados como orbitales no enlazantes.
Berilio. La configuración de Be 2 es 1 s g 2 1 s u 2 2 s g 2 s g 2 s u2 y se predice que la molécula forme una molécula de van der Waals débilmente unida como el dímero de helio.
Oxígeno. Dado que el método de determinación de configuraciones electrónicas es claro a partir de los ejemplos anteriores, consideraremos solo una molécula más en detalle, la molécula de oxígeno. Llenando los orbitales en orden de aumento de energía los dieciséis electrones de O 2 se describen por la configuración 1 s g 2 1 s u 2 2 s g 2 2 s u 2 3 s g 2 1 p u 4 1 p g 2. Las densidades orbitales se ilustran en la Figura\(\PageIndex{5}\).

Figura\(\PageIndex{5}\): Mapas de contorno de las densidades de carga orbitales moleculares para\(O_2\) a la distancia internuclear de equilibrio de 2.282 au. Solo se muestra un componente de los orbitales Ipg y 1pu. Todos los mapas son para orbitales doblemente ocupados con la excepción de aquel para 1pg para el cual cada componente del orbital doblemente degenerado contiene un solo electrón. Los nodos se indican mediante líneas discontinuas. Haga clic aquí para ver los valores de contorno.
Los orbitales moleculares de simetría p son doblemente degenerados y un conjunto lleno de orbitales p contendrá cuatro electrones. El nodo en un orbital p u está en el plano que contiene el eje internuclear y no es perpendicular a este eje como lo es el nodo en un orbital s u. (Las propiedades nodales de los orbitales se indican en la Figura\(\PageIndex{5}\)) Por lo tanto, el orbital p u se une. Un orbital p g, por otro lado, es antiadherentes porque tiene, además del nodo en el plano del eje de unión, otro en el punto medio del enlace perpendicular al eje. Los caracteres ligantes y antiadherentes de los orbitales p tienen la relación opuesta a su dependencia g y u al igual que los orbitales s.
Las densidades orbitales de 1 s g y 1 s u tienen, como en el caso de Li 2, degeneradas en distribuciones atómicas localizadas con las características de densidades de núcleo de 1 s y son no enlazantes. Los electrones de valencia de O 2 están contenidos en los orbitales restantes, característica que se refleja en la medida en que sus distribuciones de densidad se deslocalizan sobre toda la molécula. Aparte de los nodos internos que rodean los núcleos, las densidades orbitales de 2 s g y 2 s u se asemejan a la densidad de valencia de 1 s g y 1 s u distribuciones de H 2 y He 2. En la siguiente sección se presenta una discusión cuantitativa de las capacidades de unión relativa de las densidades orbitales de 2 s g, 3 s g y 1 p.
Una característica interesante de la configuración electrónica de O 2 es que su orbital exterior no está completamente ocupada. Los dos electrones p g podrían ocupar uno de los orbitales p g con espines emparejados o se les podría asignar uno a cada uno de los orbitales p g y tener espines paralelos. El principio de Hund se aplica tanto a las moléculas como a los átomos y se predice que la configuración con ocupación única de ambos orbitales p g con espines paralelos es la más estable. Esta predicción de la teoría orbital molecular respecto a la estructura electrónica de O 2 tiene una consecuencia interesante. La molécula de oxígeno debe ser magnética debido al momento angular de espín resultante que poseen los electrones. El magnetismo del O 2 se puede demostrar experimentalmente de muchas maneras, siendo una de las más simples la observación de que el oxígeno líquido es atraído hacia los polos de un imán fuerte.