
9.5: Orbitales Adhesivos y Antiadherentes
- Page ID
- 79828
- Caracterizar los orbitales moleculares ligantes y antiadherentes en\(\ce{H^{+}}\)
Los dos orbitales moleculares del\(\ce{H^{+}}\) ion se crearon a través de las combinaciones lineales de orbitales atómicos (LCaOS). La aproximación se creó a partir de la suma y la diferencia de dos orbitales atómicos. Dentro de esta aproximación, el j ésimo orbital molecular puede expresarse como una combinación lineal de muchos orbitales atómicos {\(\phi_i\)}:
\[| \psi_J \rangle = \sum_i^N c_{J,i} | \phi_i \rangle \label{9.5.12} \]
Una molécula tendrá tantos orbitales moleculares como orbitales atómicos utilizados en el conjunto de bases (\(N\)en Ecuación\(\ref{9.5.12}\)). La adición de dos orbitales atómicos corresponde a la interferencia constructiva entre dos ondas, reforzando así su intensidad; se incrementa la densidad de probabilidad de electrones internucleares. El orbital molecular correspondiente a la suma de los dos orbitales H 1 s se denomina combinación σ 1 s (partes (a) y (b) de la Figura 9.5.1 ).
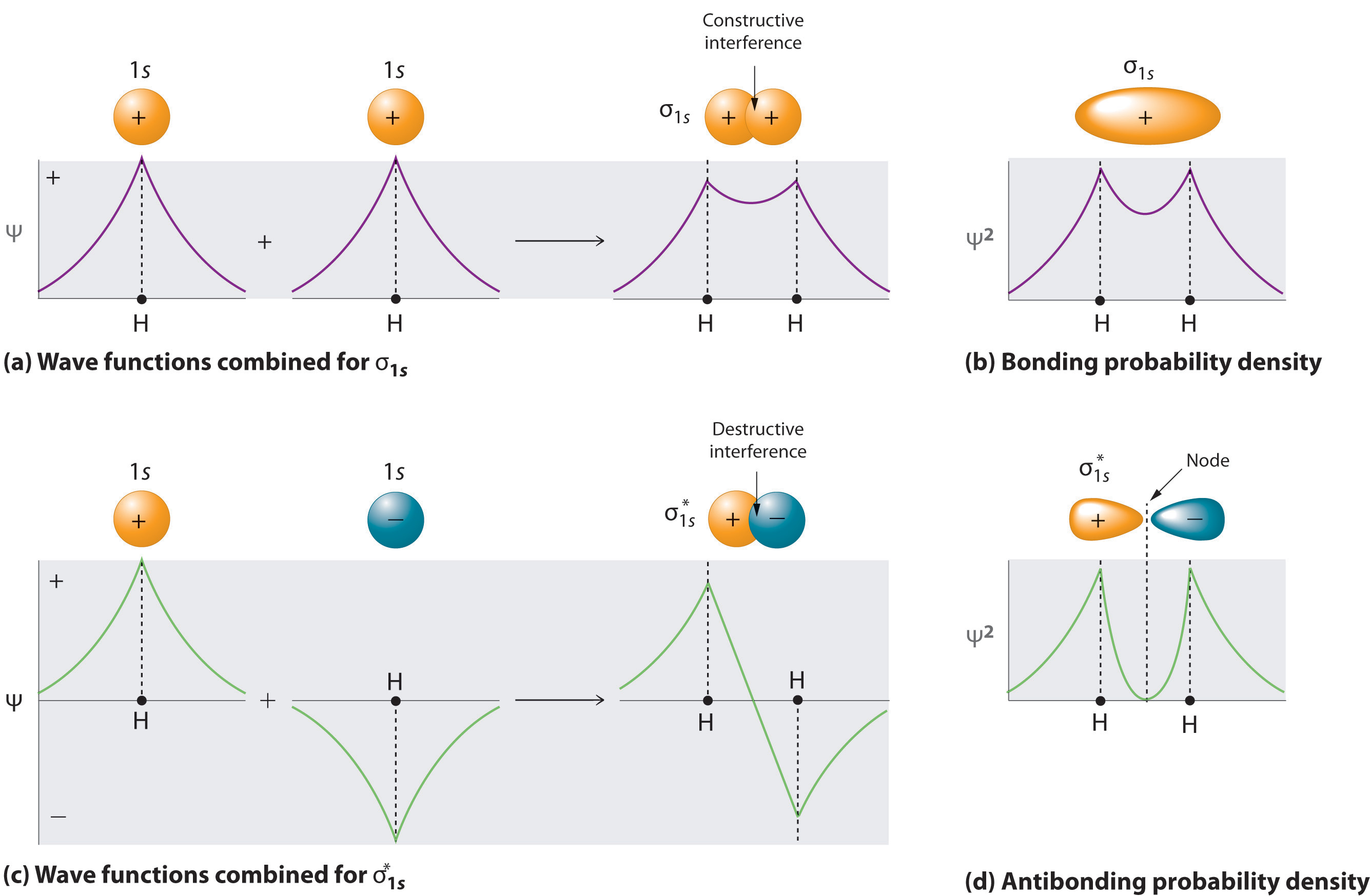
En el orbital sigma (\(σ\)), la densidad electrónica a lo largo del eje internuclear y entre los núcleos tiene simetría cilíndrica; es decir, todas las secciones transversales perpendiculares al eje internuclear son círculos. El subíndice 1 s denota los orbitales atómicos de los que se derivó el orbital molecular.
\[ | \sigma _{1s} \rangle = \dfrac{1}{\sqrt{2(1 + S )}} \left( | 1s_A \rangle + | 1s_B \rangle \right) \label{9.7.2} \]
Por el contrario, restar un orbital atómico de otro corresponde a una interferencia destructiva entre dos ondas, lo que reduce su intensidad y provoca una disminución en la densidad de probabilidad de electrones internucleares (parte (c) y parte (d) en la Figura 9.5.1 ). El patrón resultante contiene un nodo donde la densidad electrónica es cero. El orbital molecular correspondiente a la diferencia se llama\( \sigma _{1s}^{*} \) y tiene una región de probabilidad de electrones cero, un plano nodal, perpendicular al eje internuclear:
\[ | \sigma _{1s}^* \rangle = \dfrac{1}{\sqrt{2(1 - S )}} \left( | 1s_A \rangle - | 1s_B \rangle \right) \label{9.7.3} \]
La densidad de electrones en el orbital molecular σ 1 s es mayor entre los dos núcleos cargados positivamente, y las atracciones electrostáticas resultantes del electrón-núcleo reducen las repulsiones entre los núcleos. Así, el orbital σ 1 s representa un orbital molecular de unión. Un orbital molecular que se forma cuando los orbitales atómicos o lóbulos orbitales con el mismo signo interactúan para dar mayor probabilidad de electrones entre los núcleos debido al refuerzo constructivo de las funciones de onda. En contraste, los electrones en el\( \sigma _{1s}^{\star } \) orbital generalmente se encuentran en el espacio fuera de la región internuclear. Debido a que esto permite que los núcleos cargados positivamente se repelan entre sí, el\( \sigma _{1s}^{\star } \) orbital es un orbital molecular antienlace (un orbital molecular que se forma cuando los orbitales atómicos o los lóbulos orbitales de signo opuesto interactúan para dar una menor probabilidad de electrones entre los núcleos debido a refuerzo destructivo de las ondulaciones).
Los orbitales antiadherentes contienen un nodo perpendicular al eje internuclear; los orbitales de unión no.
Debido a que los electrones en el orbital σ 1 s interactúan simultáneamente con ambos núcleos, tienen una energía menor que los electrones que interactúan con un solo núcleo. Esto significa que el orbital molecular σ 1 s tiene una energía menor que cualquiera de los orbitales atómicos de hidrógeno 1 s. Por el contrario, los electrones en el\( \sigma _{1s}^{\star } \) orbital interactúan con un solo núcleo de hidrógeno a la vez. Además, están más lejos del núcleo de lo que estaban en los orbitales atómicos de hidrógeno progenitor 1 s. En consecuencia, el orbital\( \sigma _{1s}^{\star } \) molecular tiene una energía mayor que cualquiera de los orbitales atómicos de hidrógeno 1 s. El orbital molecular σ 1 s (enlace) se estabiliza en relación con los orbitales atómicos de 1 s, y el orbital molecular\( \sigma _{1s}^{\star } \) (antienlace) se desestabiliza. Los niveles de energía relativa de estos orbitales se muestran en el diagrama de niveles de energía (un dibujo esquemático que compara las energías de los orbitales moleculares (enlace, antienlace y no unión) con las energías de los orbitales atómicos padres) en la Figura 9.5.2
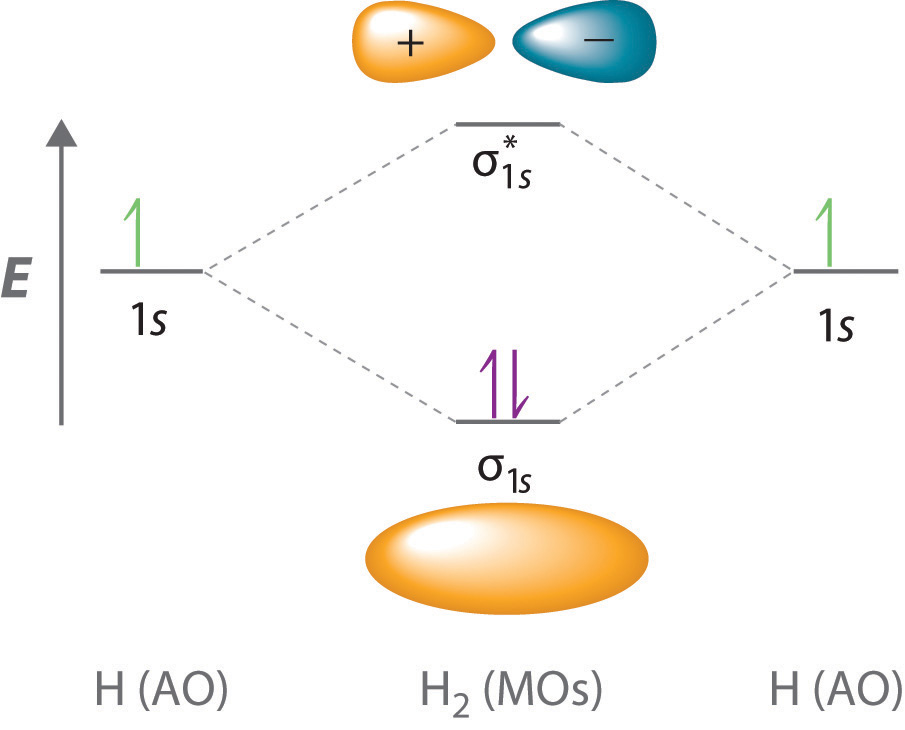
Un orbital molecular de unión es siempre más bajo en energía (más estable) que los orbitales atómicos componentes, mientras que un orbital molecular antienlace es siempre mayor en energía (menos estable).
Expansión más allá del conjunto de bases orbitales 1s
Esta imagen de unión en H 2 + en la sección anterior es muy simple, pero da resultados razonables cuando se compara con un cálculo exacto. La distancia de enlace de equilibrio es de 134pm en comparación con 106pm (exacta), y una energía de disociación es de 1.8 eV en comparación con 2.8 eV (exacta). Para describir mejor la unión química necesitamos tener en cuenta el aumento en la densidad de electrones entre los dos núcleos. Los orbitales 1s por sí solos no son particularmente buenos para este propósito porque son esféricamente simétricos y no muestran preferencia por el espacio entre los núcleos atómicos. El uso de orbitales atómicos adicionales puede corregir esta situación y proporcionar parámetros adicionales, los cuales pueden ser optimizados por el método variacional lineal, para dar una mejor función con una menor energía y una descripción más precisa de la densidad de carga.
La energía del orbital molecular no normalizado puede calcularse a partir del valor de expectativa integral del Hamiltoniano,
\[E_{J} = \dfrac{\left \langle \psi _{J} | \hat {H} _{elec} | \psi _{J} \right \rangle}{\left \langle \psi _{J} | \psi _{J} \right \rangle} \label {9.5.13} \]
Esta es la energía variacional\(| \psi _{J} \rangle\) que se utiliza como función de onda del sendero. Después de sustituir la expansión LCAO por\(| \psi _{J} \rangle\) (Ecuación\ ref {9.5.12}) en la expresión energética de la Ecuación\ ref {9.5.13} da como resultado:
\[\begin{align} E_{J} &= \dfrac{\left \langle \displaystyle \sum_i c_{J,i}^* \phi_i \right | \hat {H} _{elec} \left | \displaystyle \sum_j c_{J,i} \phi_j \right \rangle}{\left \langle \displaystyle \sum_i c_{J,i}^* \phi_j | \displaystyle \sum_j c_{J,j} \phi_j\right \rangle} \label {9.5.14} \\[4pt] &= \dfrac{ \displaystyle \sum_{i,j} c_{J,i}^* c_{J,j} \left \langle \phi_i \right| \hat {H} _{elec} \left| \phi_j \right \rangle}{ \displaystyle\sum_{i,j} c_{J,i}^* c_{J,j} \left \langle \phi_i | \phi_j\right \rangle} \label {9.5.15} \\[4pt] &= \dfrac{ \displaystyle \sum_{i,j} c_{J,i}^* c_{J,j} H_{ij}}{ \displaystyle \sum_{i,j} c_{J,i}^* c_{J,j} S_{ij} } \label {9.5.16} \end{align} \]
donde\(H_{ij}\) está el elemento matriz hamiltoniano.
\[H_{ij} = \langle \phi_i | \hat {H} _{elec} | \phi_j \rangle \nonumber \]
Siguiendo el teorema variacional, para determinar los coeficientes de la expansión de LCAO\(c_i\), necesitamos minimizar\(E_J\)
\[ \dfrac{\partial E_J}{\partial c_k} = 0 \label{9.5.17} \]
para todos\(k\). Esto requiere resolver ecuaciones\(N\) lineales para que se mantengan verdaderas (donde\(N\) está el número de orbitales atómicos en la base)
\[ \sum_{i=1}^{N} c_i (H_{ki} - ES_{ki}) = 0 \label{9.5.18} \]
Estas ecuaciones son las ecuaciones seculares y se discutieron previamente en el contexto de la aproximación del método variacional lineal. Para la expansión de dos conjuntos básicos (\(N\)) en la Figura 9.5.1 , estos son
\[\begin{array}{rcl} c_1(H_{11} - ES_{11}) + c_2(H_{12} - ES_{12}) & = & 0 \\ c_1(H_{12} - ES_{12}) + c_2(H_{22} - ES_{22}) & = & 0 \end{array} \label{9.5.19} \]
donde\(c_1\) and \(c_2\) are the coefficients in the linear combination of the atomic los orbitales utilizados para construir el orbital molecular. Escribir este conjunto de ecuaciones lineales homogéneas en forma de matriz da
\[\begin{pmatrix} H_{11} - ES_{11} & H_{12} - ES_{12} \\ H_{12} - ES_{12} & H_{22} - ES_{22} \end{pmatrix} \begin{pmatrix} c_1 \\ c_2 \end{pmatrix} = \begin{pmatrix} 0 \\ 0 \end{pmatrix} \label{9.5.20} \]
Resolver estas ecuaciones seculares con N orbitales atómicos diferentes en la expansión (Ecuación\(\ref{9.5.12}\)) requiere encontrar las raíces N de un polinomio de orden N.
\[\left|\begin{array}{lcc} H_{11} - ES_{11} & H_{12} - ES_{12} & \ldots\\ H_{12} - ES_{12} & H_{22} - ES_{22} &\ldots \\ \ldots &\ldots &\ldots \end{array}\right|=0\label{23} \]
Cada orbital molecular (\(| \psi_J \rangle\)) de este tratamiento tiene una energía\(E_J\) que viene dada por un conjunto diferente de coeficientes,\(\{c_{ij}\}\) donde\(i\) recorre todas\(N\) las funciones en la base (es decir, el número de los orbitales atómicos en la aproximación LCAO de la Ecuación\(\ref{9.5.12}\)), y \(J\)corre sobre orbitales moleculares. Resuelve el conjunto de ecuaciones lineales usando esa específica\(E_J\) para determinar\(c_{ij}\) valores.
- Seleccionar un conjunto de N funciones básicas
- Determine todos los valores N (N —1) /2 de ambos\(H_{ij}\) y\(S_{ij}\)
- Formar el determinante secular; determinar las raíces N\(E_j\) de la ecuación secular
- Para cada\(E_J\) resolver el conjunto de ecuaciones lineales para determinar los coeficientes del conjunto de bases\ (c_ {ij\}) para el j-ésimo orbital molecular
Para más información sobre cómo resolver las ecuaciones seculares consulta aquí.
Cuanto mayor sea el número de orbitales atómicos\(N\) que se combinan para generar los orbitales moleculares (Ecuación\(\ref{9.5.12}\)), más precisa es la aproximación de LCAO. Esto se espera a partir de nuestras discusiones sobre los ejemplos del método variacional. Por lo tanto, los orbitales\(\psi_-\) moleculares\(\psi_+\) y para\(H_2^+\) se expresan mejor con funciones de onda hidrogénicas de mayor energía
\[| \psi_J \rangle = c_{J,1} 1s_A + c_{J,2} 1s_B + c_{J,3} 2s_A + c_{J,4} 2s_B + c_{J,5} 2p_{z,A} + c_{J,6} 2p_{z,B} \label{9.5.24} \]
Las razones por las que solo se incluyen los orbitales\(p_z\) atómicos en esta expansión se discuten más adelante.
Colaboradores y Atribuciones
Adapted from "Quantum States of Atoms and Molecules" by David M. Hanson, Erica Harvey, Robert Sweeney, Theresa Julia Zielinski