
23.1: Un diagrama de fases resume el comportamiento sólido-líquido-gas de una sustancia
- Page ID
- 79631
Un buen mapa te llevará a tu destino con facilidad, siempre que sepas leerlo. Un mapa es un ejemplo de diagrama, una representación pictórica de un cuerpo de conocimiento. En la ciencia juegan un papel considerable. Junto a las parcelas y tablas, los diagramas son un medio importante para hacer accesible la información y/o el conocimiento teórico.
Construirlos requiere un poco de pensamiento. Quieres representar tanto de lo que conoces y dar una imagen tan precisa de ello sin transmitir nada incorrecto. Si el dibujo se puede hacer a escala eso lo hace bastante más potente, pero esto no es estrictamente necesario. Se necesita dar una observación como no escalar o esquemáticamente si corresponde. Un buen subtítulo o descripción es esencial.
Estabilidad Termodinámica y Fluctuaciones
Existen diferentes tipos de equilibrio, además del equilibrio estable que representa un mínimo absoluto en la\(G\) función. Por supuesto\(G\) es potencialmente una función de un gran número de variables, pero veamos un diagrama en el que\(G\) se muestra como una función de una sola variable no especificada. Se podría pensar en la densidad, la fracción molar de uno de los componentes de una mezcla o un campo eléctrico aplicado o lo que sea, pero el argumento es general.
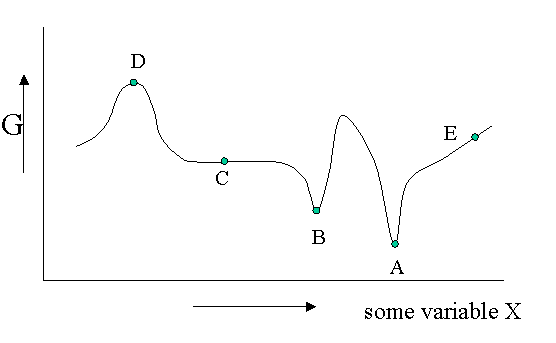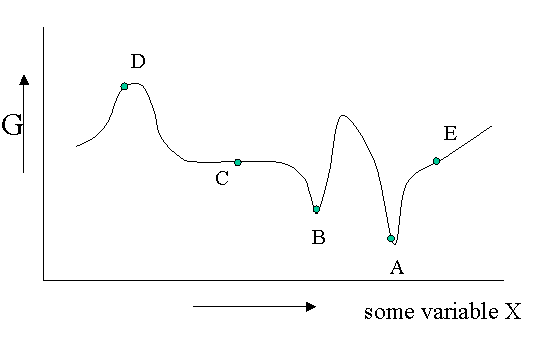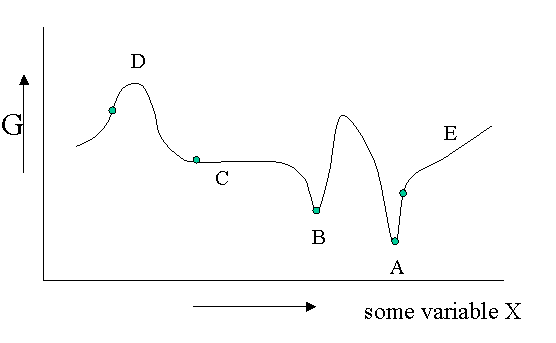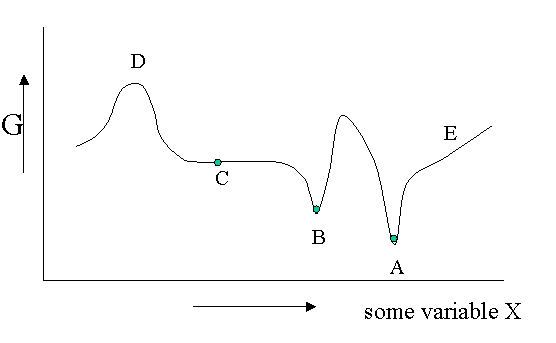
La Figura 23.1.1 es útil para señalar que además de un equilibrio estable (\(A\)) también puede haber un equilibrio metaestable (B) o un equilibrio indiferente (\(C\)). Las derivadas locales de\(G\) (versus todas las variables de las cuales solo mostramos una) son cero en los tres casos, lo que significa que los cambios en las variables no son espontáneos. Para un equilibrio lábil (\(D\)) lo contrario es cierto. Cualquier pequeña desviación hará que el sistema funcione cuesta abajo. (Tenga en cuenta que la segunda derivada tiene el signo opuesto en comparación con cas A) y B)) Un equilibrio lábil rara vez se observa o nunca excepto en un circo donde los artistas se deleitan en equilibrar objetos en sus cabezas (porque pagas por ello).. Esto suele requerir pequeñas correcciones continuas para mantener el precario equilibrio. Todos los demás puntos de nuestro diagrama representan el estado de inestabilidad porque localmente no\(dG\) es cero y puede tener lugar un proceso espontáneo.
El hecho de que\(dG=0\) en los puntos de equilibrio no signifique pequeñas desviaciones del mínimo no puede ocurrir a veces. Hemos visto, por ejemplo, que la distribución de Boltzmann era simplemente la distribución más probable. El más probable es el que tiene el mayor número de realizaciones W. Otra forma de decir eso es que es el que tiene la mayor entropía S. Una distribución ligeramente menos probable puede ocurrir de vez en cuando por casualidad. Tendrá un poco menos de entropía, pero igual\(\langle E \rangle\). Eso significa que tendrá un poco más alto\(G\) (\(G=H-TS\)). De vez en cuando por lo tanto\(G\) fluctuará un poco. Tales fluctuaciones son muy pequeñas para sistemas grandes, pero son de mayor importancia relativa para sistemas pequeños (como una nanopartícula). (El promedio estadístico funciona mejor en conjuntos grandes.)
Las fluctuaciones en\(G\) significan que también pueden ocurrir pequeñas fluctuaciones en sus variables como densidad, etc. Suelen mantenerse bajo control, porque ya no\(dG\) es cero cuando se aleja del estado de equilibrio. Esto hace que el sistema vuelva al mínimo espontáneamente. Podrías imaginarte el sistema tambaleándose un poco en su pozo G. Esto se mantiene para equilibrios estables y metaestables por igual.
En el caso indiferente (\(C\)) sin embargo la derivada es cero (o muy cercana a cero) para un rango de valores vecinos de alguna variable. En contraste con\(A\) y\(B\) también la segunda derivada es cero. Esto significa que hay poca penalización a desviaciones mucho mayores en la variable. Si esta variable es la densidad el sistema se vuelve lechoso y muestra opalescencia una fuerte dispersión de la luz porque el índice de refracción depende de la densidad fuertemente fluctuante. Esto se observa cerca de puntos críticos y se denomina opalescencia crítica.
Diagramas de fase unarios
Un diagrama de fases unario resume los estados de equilibrio de una sola sustancia pura. Veremos que también podemos mirar mezclas de dos componentes (diagramas binarios) o más (ternario, cuaternario, quinario, senario etc.). Por lo general, un diagrama de fases solo mapea equilibrios estables, pero ocasionalmente se pueden dar también los metaestables (por ejemplo, con una línea discontinua).
Curva de equilibrio líquido-vapor
Hemos visto que la función de Gibbs\(G\) depende fuertemente (logarítmicamente) de la presión para un gas, pero sólo ligeramente (y linealmente) para un líquido. Las dos curvas se cruzan en un punto que representa la presión de vapor de equilibrio del líquido. A presiones más bajas el vapor es más estable, a las más altas el líquido. (Para un sólido las mismas bodegas que para el líquido). Esto quiere decir que salvo en el punto de intersección solo observaremos una fase.
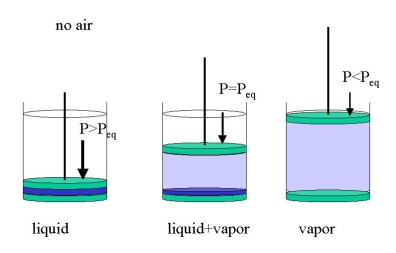
Es importante destacar que esto se sostiene en ausencia de otra materia, por ejemplo, cuando ponemos solo agua en un cilindro evacuado (Figura 23.1.2 ). Podemos obtener tres casos:
- comprimimos el cilindro hasta que solo contenga el líquido bajo presión hidrostática (\(P>P_{eq}\))
- expandimos el cilindro hasta que toda el agua se haya vaporizado (\(P<p_{eq}\)) >
- dejamos que parte del agua se evapore, lo suficiente para que el espacio sobre el líquido se llene con una presión de vapor de equilibrio (\(P=P_{eq}\))
A temperatura ambiente\(P_{eq}\) para el agua es de sólo unos 15 Torr. Si aplicamos 1 bar -o dejamos que la atmósfera haga el trabajo- solo tendremos agua líquida. Si hay otros gases presentes, por ejemplo, aire, debemos distinguir entre la presión total (por ejemplo, 1 bar) y la presión de vapor de equilibrio que ahora será la presión parcial. En un cilindro con agua y una barra de aire apenas el agua suficiente se evaporará para establecer el equilibrio. La evaporación se limitará a la interfaz gas-líquido a menos que la presión parcial sea igual a la presión total. Entonces el líquido hervirá.
(Sin embargo, permiten que el volumen se expanda., ¿por qué? Si el volumen es constante la presión se acumula y la ebullición se detendrá.
Si consideramos el conjunto de presiones de equilibrio como una función de la temperatura y trazamos que en un diagrama P vs T tenemos un componente de nuestro diagrama de fases.
Curva de equilibrio gas-sólido
Para los sólidos la situación es similar ya que la\(G(P)\) curva vuelve a ser una línea recta casi plana. La intersección con la curva logarítmica para el gas definirá una presión de equilibrio para la coexistencia gas-sólido. Generalmente las presiones de vapor por encima de los sólidos son bastante pequeñas, pero no despreciables. En cuanto a los líquidos podemos construir una línea que represente las presiones de equilibrio para sublimación en función de la temperatura y agregarla al diagrama de fases.
Curva de equilibrio líquido-sólido
El\(\text{solid} \rightleftharpoons \text{liquid}\) equilibrio se conoce como fusión o congelación no depende mucho de la presión. Por lo general, los puntos de fusión aumentan un poco con la presión, aunque el agua es una excepción peculiar. Se expande al congelarse y el punto de fusión baja (un poco) con la presión. En nuestro diagrama esto representará una línea casi vertical inclinada un poco hacia adelante para la mayoría de las sustancias, pero hacia atrás para el agua y algunas otras.
Armando la curva
Las tres líneas se juntan en el punto triple, el único punto donde las tres fases están en equilibrio entre sí. Para el agua, su temperatura es solo 0.01 K diferente del punto de fusión normal (273.16 K) y su presión es de solo 4.58 Torr. Los puntos de intersección con una línea que representa la presión atmosférica dan los puntos de fusión y ebullición a esa presión.
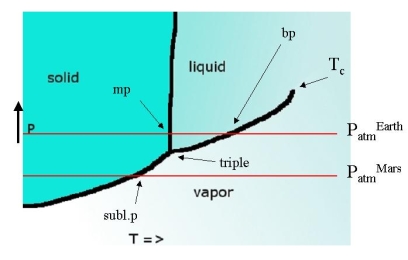
Si el punto triple se encuentra por encima de la línea que representa la presión atmosférica esto implica que nunca se observa un líquido. En la tierra el CO 2 es tal sustancia. La intersección de la línea de equilibrio sólido-vapor con la línea de 1 bar representa un estado en el que el sólido 'hervirá' (se evaporará de adentro hacia afuera). Esto se conoce como el punto de sublimación. Los puntos de fusión a P=1 bar se conocen como el punto de fusión estándar, el único ligeramente diferente a 760 Torr = 1 atm se llama punto de fusión normal. Lo mismo ocurre con los puntos de ebullición y sublimación.
No hay nada mágico en ello\(P=1 \,bar\). Simplemente pasa a ser la presión de nuestro planeta natal. En un planeta con presiones atmosféricas más altas bien\(CO_2\) puede ser un líquido y en tal planeta, todos los puntos de ebullición serán bastante diferentes (más altos que en la tierra). Los puntos de fusión también diferirán, pero sólo ligeramente así. En Marte donde la presión atmosférica es mucho menor el agua no puede ocurrir en forma líquida, al igual que el dióxido de carbono en la tierra - sublima.
También debemos darnos cuenta de que en un recipiente cerrado (ampolla de vidrio, sartén DSC herméticamente sellada), podemos observar puntos de fusión a valores de temperatura solo muy ligeramente diferentes, pero no veremos un efecto de ebullición. ¿Por qué?
Para ver un punto de ebullición el recipiente debe estar abierto a la (¡constante! ) 1 bar de presión de la atmósfera terrestre que lo define y provoca el fenómeno de ebullición. Si la ampolla está sellada generará su propia presión (autógena), dependiendo de lo que le pongas, cuánto de ella en relación con el volumen, qué tan volátil es y la temperatura. La presión autógena no interfiere mucho con el punto de fusión (la línea de fusión es casi vertical), pero como\(P\) cambia con la temperatura es posible que nunca alcances condiciones de ebullición.
En experimentos DSC es posible observar puntos de ebullición solo si la sartén ha sido cuidadosamente perforada con un agujero de tamaño conocido. Debe ser lo suficientemente grande como para que la presión dentro de la sartén no se acumule por encima de la atmosférica, sino lo suficientemente pequeña como para que no cause pérdida prematura de masa durante la carrera. Este último estropea el cálculo del valor intensivo (por mol, por gramo) del calor de vaporización.
La línea de evaporación del líquido termina en un punto que nos hemos encontrado antes: el punto crítico\(T_C\). A medida que aumenta la temperatura, las fases líquida y vapor en equilibrio entre sí comienzan a parecerse cada vez más y en\(T_c\) ellas se unen. En este punto el equilibrio líquido-gas se vuelve indiferente con respecto a la densidad y se producen grandes fluctuaciones que conducen a la opalescencia crítica.
Observe que existe una relación de dimensionalidad entre los objetos del diagrama y el número de fases presentes:
- 2 planos D: una fase
- Curvas 1 D: dos fases
- Punto 0 D: tres fases
Como ves la suma es siempre tres.
Número de moles
Hasta ahora hemos considerado típicamente una sustancia a la vez, pero para los químicos es imperativo tratar con más de una porque normalmente estamos cambiando una por otra en nuestras reacciones. Esto significa que el número de moles\(n\), que a menudo simplemente establecemos igual a uno ahora se convierte en una variable importante por derecho propio. Además en realidad tendremos dos (o más) de ellos: el número de moles del componente uno y el uno para el otro componente. Esto hace que n sea una variable mucho menos trivial.
Esto ya es el caso en un simple punto de fusión, digamos cuando el hielo se derrite, porque nos estamos ocupando de cambiar las cantidades de hielo y agua:
\[n_{ice} + n_{water} = n_{total} \nonumber \]
Si todo lo que hacemos es convertir el agua en hielo o viceversa, tenemos\(dn_{total}=0\), para que:
\[dn_{ice} =- dn_{water} \nonumber \]
Para lidiar con el cambio de n, necesitamos expandir un poco nuestra notación matemática.
Variables parciales
Hasta ahora simplemente hemos dividido nuestras funciones termodinámicas si eran extensas por el número de moles y llegaban a valores molares intensivos:
\[G_{molar} = \dfrac{G}{ n} \nonumber \]
\[V_{molar} = \dfrac{V}{n} \nonumber \]
Hemos escrito valores molares tan intensivos escribiendo una barra sobre el símbolo G o V. Debemos señalar que escalar la función de esta manera se aleja de la suposición de que la función\(G\) depende de la variable n como una línea recta que pasa por el origen.
Si tenemos el mismo compuesto puro en dos fases, como el hielo y el agua todavía podemos aplicar este principio y escribir:
\[G_{molar}^{ice} = \dfrac{G^{ice}}{n^{ice}} \nonumber \]
\[V_{molar}^{ice} = \dfrac{V^{ice}}{n^{ice}} \nonumber \]
\[G_{molar}^{water} = \dfrac{G^{water}}{n^{water}} \nonumber \]
\[V_{molar}^{water} = \dfrac{V^{water}}{n^{water}} \nonumber \]
Si tenemos una mezcla de dos sustancias presentes como\(n_1\) y\(n_2\) moles la dependencia no necesita ser lineal en cualquiera de si las dos sustancias interactúan entre sí. Esto también es cierto para funciones como el volumen de una mezcla líquida. En presencia de interacciones los volúmenes no tienen que ser linealmente aditivos. Podemos definir un valor molar parcial de, por ejemplo, para el volumen:
\[V_{partial molar,1} = \dfrac{\partial V}{\partial n_1} \nonumber \]
a\(n_2\) = constante
\[V_{partial molar,2} = \dfrac{\partial V}{\partial n_2} \nonumber \]
a\(n_1\) = constante
La notación de poner una barra sobre el\(V\) símbolo también se usa para estas cantidades parciales. Se han medido volúmenes molares parciales para muchos sistemas binarios. Son funciones de la composición (fracción molar) así como de la temperatura y en menor medida la presión.
La energía parcial molar libre de Gibbs (\(\left(\dfrac{∂G}{∂n_i}\right)_{P,T}\), todas las demás n) se denota con\(μ\) y se llama el potencial termodinámico.
Cuando los números de moles pueden cambiar podemos escribir el cambio correspondiente en\(G\) como:
\[dG = -SdT + VdP + \sum_i^N \left( \dfrac{\partial G}{\partial n_1} \right)_{P,T,n_{j\neq i}}dn_i \nonumber \]
\[dG = -SdT + VdP + \sum_i^N μ_idn_i \nonumber \]
sobre\(N\) fases en el sistema. Como puede ver estamos agregando un conjunto de variables conjugadas\(μ_in_i\) para cada fase\(i\). Si estamos considerando un componente puro (pero en diferentes modificaciones, como hielo y vapor), aún podemos escribir:
\[ μ_i = \left( \dfrac{\partial G}{\partial n_1} \right)_{P,T,n_{j\neq i}} = \dfrac{G_i}{n_i} \nonumber \]
Tan pronto como estamos tratando con mezclas realmente tenemos derivados.