
5.3: Cambio Químico
- Page ID
- 70828

Sondas Experimentales de Cambio Químico
Muchas de las mismas herramientas que se utilizan para determinar las estructuras de las moléculas también se pueden utilizar para seguir los cambios que sufre la molécula ya que está involucrada en una reacción química. Específicamente, para cualquier reacción en la que un tipo de molécula\(A\) se convierta en otro tipo\(B\), es necesario tener
- la capacidad de identificar, a través de alguna medición física, las firmas experimentales de ambos\(A\) y\(B\),
- la capacidad de relacionar la magnitud de estas señales experimentales con las concentraciones\([A]\) y\([B]\) de estas moléculas, y
- la capacidad de monitorear estas señales como funciones del tiempo para que estas concentraciones puedan ser seguidas a medida que evoluciona el tiempo.
El tercer requisito es lo que permite determinar las velocidades a las que reaccionan\(B\) las moléculas\(A\) y.
Muchas de las herramientas experimentales utilizadas para identificar moléculas (por ejemplo, la RMN permite identificar grupos funcionales y grupos funcionales cercanos al vecino, IR también permite ver grupos funcionales) y para determinar sus concentraciones tienen escalas de tiempo restringidas sobre las cuales pueden ser utilizadas. Por ejemplo, los espectros de RMN requieren que la muestra se estudie durante aproximadamente 1 segundo o más para obtener una señal utilizable. Asimismo, un análisis espectroscópico de masas de una mezcla de especies reaccionantes puede requerir muchos segundos o minutos para llevar a cabo. Estas restricciones, a su vez, limitan las tasas de reacciones que se pueden seguir utilizando estas herramientas experimentales (por ejemplo, no se puede usar RMN de espectroscopía de masas para seguir una reacción que ocurre en una escala de tiempo de\(10^{-12}\) s).
Especialmente para reacciones muy rápidas y para reacciones que involucran especies inestables que no pueden manejarse fácilmente, a menudo se utilizan los llamados enfoques experimentales bomba-sonda.
Por ejemplo, supongamos que uno estuviera interesado en estudiar la reacción de\(Cl\) los radicales (por ejemplo, como se forman en la descomposición de cloroflurocarbonos (CFC) por luz ultravioleta) con el ozono para generar\(ClO\) y\(O_2\):
\[Cl + O_3 \rightarrow ClO + O_2^. \tag{5.3.1}\]
No se puede simplemente depositar una cantidad conocida de\(Cl\) radicales de un recipiente en un recipiente en el que se haya preparado un gas\(O_3\) de concentración conocida; los\(Cl\) radicales se recombinarán y reaccionarán con otras especies, dificultando su determinación de sus concentraciones. Entonces, alternativamente, se colocan concentraciones conocidas de algún precursor\(Cl\) radical (por ejemplo, un CFC o alguna otra especie X-Cl) y ozono en un recipiente de reacción. Luego se usa, por ejemplo, un pulso de luz muy corto cuyas frecuencias de fotones están sintonizadas a una transición que hará que el precursor X-Cl experimente una rápida fotodisociación:
\[h\nu + X-Cl \rightarrow X + Cl^.\tag{5.3.2}\]
Debido a que la fuente de luz de bombeo utilizada para preparar los\(Cl\) radicales es de muy corta duración (\(\Delta{t}\)) y debido a que la disociación X-Cl es pronta, se sabe\(\Delta{t}\), dentro, el tiempo en el que los radicales Cl comienzan a reaccionar con el ozono. La concentración inicial de los\(Cl\) radicales se puede conocer si se conoce el rendimiento cuántico para la\(h\nu + X-Cl \rightarrow X + Cl\) reacción, esto significa que se debe conocer la intensidad de los fotones, la probabilidad de absorción de fotones por X-Cl, y la fracción de moléculas X-Cl excitadas\(X + Cl\) que se disocian para producir. Dicha información está disponible (aunque de estudios anteriores bastante tediosos) para una variedad de precursores X-Cl.
Entonces, conociendo el momento en que se forman los\(Cl\) radicales y sus concentraciones iniciales, se permite entonces que la\(Cl + O_3 h\nu \rightarrow ClO + O_2\) reacción continúe por algún tiempo de duración\(\Delta{t}\). Uno entonces, at\(t =\Delta{t}\), usa una segunda fuente de luz para sondear ya sea la concentración del\(ClO\), el\(O_2\) o el\(O_3\), para determinar el grado de progreso de la reacción. Qué especies se monitorean así depende de la disponibilidad de fuentes de luz cuyas frecuencias absorban estas especies. Dichos experimentos de sonda se llevan a cabo en una serie de retardos de tiempo\(\Delta{t}\), cuyo resultado es la determinación de las concentraciones de algún producto o especie reaccionante en diversos momentos después de que el evento de bombeo inicial creó los\(Cl\) radicales reactivos. De esta manera, se puede monitorear, por ejemplo, la\(ClO\) concentración en función del tiempo después de que el\(Cl\) comience a reaccionar con el\(O_3\). Si uno tiene razones para creer que la reacción ocurre en un solo evento bimolecular como
\[Cl + O_3 \rightarrow ClO + O_2 \tag{5.3.3}\]
entonces se puede extraer la constante de velocidad k para la reacción usando el siguiente esquema cinético;
\[\dfrac{d[ClO]}{dt} = k [Cl] [O_3].\tag{5.3.4}\]
Si la concentración inicial de\(O_3\) es grande en comparación con la cantidad de\(Cl\) que se forma en el evento de bombeo, se\([O_3]\) puede tomar como constante y conocida. Si\(Cl\) se denota la concentración inicial de\([Cl]_0\), y\(ClO\) se llama la concentración de\(x\), esta ecuación cinética se reduce a
\[\dfrac{dx}{dt} = k ( [Cl]_0 -x) [O_3]\tag{5.3.5}\]
cuya solución es
\[[ClO] = x = [Cl]_0 (1 - \exp(-k[O_3]t)).\tag{5.3.6}\]
Entonces, conociendo la\([ClO]\) concentración en función del retardo de tiempo\(t\), y conociendo la concentración inicial de ozono así\([O_3]\) como la concentración inicial de\(Cl\) radicales, se puede encontrar la constante de velocidad\(k\).
Dichos experimentos bomba-sonda son necesarios cuando se quiere estudiar especies que deben generarse y dejarse reaccionar inmediatamente. Esto es esencialmente siempre el caso cuando uno o más de los reactivos es una especie altamente reactiva como un radical. Existe otro tipo de experimento que puede usarse para sondear reacciones muy rápidas si la reacción y su reacción inversa pueden ponerse en equilibrio en la medida en que los reactivos y productos existan en concentraciones medibles. Por ejemplo, considere la reacción de una enzima E y un sustrato S para formar el complejo enzima-sustrato ES:
\[E + S \rightleftharpoons ES.\tag{5.3.7}\]
En equilibrio, la tasa de avance
\[k_f = [E]_{eq} [S]_{eq} \tag{5.3.8}\]
y la tasa inversa
\[k_r = [ES]_{eq} \tag{5.3.9}\]
son iguales:
\[k_f [E]_{eq} [S]_{eq} = k_r [ES]_{eq} \tag{5.3.10}\]
La idea detrás de las llamadas técnicas de perturbación es comenzar con una reacción que se encuentra en tal condición de equilibrio y luego usar algunos medios externos para perturbar ligeramente el equilibrio. Debido a que se supone que tanto las tasas de avance como de retroceso son muy rápidas, es esencial utilizar una perturbación que pueda alterar las concentraciones muy rápidamente. Esto normalmente impide simplemente añadir una pequeña cantidad de una o más de las especies reaccionantes al recipiente de reacción. En cambio, uno podría emplear, por ejemplo, una fuente de luz rápida o un pulso de campo eléctrico para perturbar el equilibrio hacia un lado u otro. Por ejemplo, si se conoce la termoquímica de reacción, la constante de equilibrio se\(K_{eq}\) puede cambiar calentando rápidamente la muestra (por ejemplo, con un pulso láser rápido que se absorbe y calienta rápidamente la muestra) y usando
\[\dfrac{d \ln{K_{eq}}}{dT} = \dfrac{\Delta{H}}{RT^2} \tag{5.3.11}\]
para calcular el cambio en\(K_{eq}\) y por lo tanto los cambios en las concentraciones causados por el calentamiento repentino. Alternativamente, si la polaridad de los reactivos y productos es sustancialmente diferente, se puede usar un campo eléctrico aplicado rápidamente para cambiar rápidamente las concentraciones del reactivo y las especies de productos.
En tales experimentos, las concentraciones de la especie se desplazan en una pequeña cantidad\(\delta\) como resultado de la aplicación de la perturbación, de manera que
\[[ES] = [ES]_{eq} - \delta \tag{5.3.12}\]
\[[E] = [E]_{eq} + \delta \tag{5.3.13}\]
\[[S] = [S]_{eq} + \delta \tag{5.3.14}\]
una vez que la perturbación ha sido aplicada y luego apagada. Posteriormente, la siguiente ley de tasas regirá la evolución temporal del cambio de concentración d:
\[- \dfrac{d\delta}{dt} = - k_r ([ES]_{eq} -\delta) + k_f ([E]_{eq} + \delta) ([S]_{eq} + \delta). \tag{5.3.15}\]
Suponiendo que eso\(\delta\) es muy pequeño (para que se descuide el término que implica\(\delta^2\) cam) y utilizando el hecho de que las tasas hacia adelante y hacia atrás se equilibran en equilibrio, esta ecuación para la evolución temporal de\(\delta\) puede reducirse a:
\[- \dfrac{d\delta}{dt} = (k_r + k_f [S]_{eq} + k_f [E_{eq}]) d. v \tag{5.3.16}\]
Entonces, las desviaciones de concentración del equilibrio volverán al equilibrio (es decir,\(\delta\) disminuirán a cero) exponencialmente con un coeficiente de tasa efectivo que es igual a una suma de términos:
\[k_{eff} = k_r + k_f [S]_{eq} + k_f [E_{eq}] \tag{5.3.17}\]
involucrando tanto las constantes de tasa hacia adelante como hacia atrás.
Entonces, al perturbar rápidamente una mezcla de reacción en equilibrio por un corto período de tiempo y posteriormente siguiendo las concentraciones de los reactivos o productos a medida que regresan a sus valores de equilibrio, se puede extraer el coeficiente de tasa efectiva\(k_{eff}\). Haciendo esto a una variedad de diferentes concentraciones iniciales de equilibrio (e, g.,\([S]_{eq}\) y\([E]_{eq}\)), y viendo cómo\(k_{eff}\) cambia, se pueden determinar las constantes de velocidad tanto hacia adelante como hacia atrás.
Tanto los métodos bomba-sonda como los métodos de perturbación requieren que uno sea capaz de crear rápidamente (o perturbar) concentraciones de especies reactivas y que se tenga disponible una sonda experimental que permita seguir las concentraciones de al menos algunas de las especies a medida que evoluciona el tiempo. Claramente, para reacciones muy rápidas, esto significa que se deben usar herramientas experimentales que puedan responder en una escala de tiempo muy corta. La tecnología láser moderna y los métodos de haz molecular han proporcionado algunas de las herramientas más utilizadas. Estos enfoques experimentales se discuten con cierto detalle en el Capítulo 8.
Simulación Teórica de Cambio Químico
El enfoque teórico más común para simular una reacción química es utilizar la dinámica newtoniana para seguir el movimiento de los núcleos en una superficie de energía electrónica Born-Oppenheimer. Si la molécula de interés contiene pocos (\(N\)) átomos, dicha superficie podría calcularse (utilizando los métodos discutidos en el Capítulo 6) en un gran número de geometrías moleculares\(\{Q_K\}\) y luego ajustarse a una función analítica\(E(\{q_J\})\) de la\(3N-6\) o\(3N-5\) variables denotadas\(\{q_J\}\). Conociendo\(E\) como una función de estas variables, uno puede entonces calcular las fuerzas
\[F_J = -\dfrac{\partial{E}}{\partial{q_J}} \tag{5.3.18}\]
a lo largo de cada coordenada, y luego usar las ecuaciones de Newton
\[m_J \dfrac{d^2q_J}{dt^2} = F_J \tag{5.3.19}\]
para seguir la evolución temporal de estas coordenadas y de ahí el avance de la reacción. Los valores de las coordenadas\(\{q_J(t_L)\}\) en una serie de tiempos discretos\(t_L\) constituyen lo que se denomina trayectoria clásica. Para simular una reacción química, se inicia la trayectoria con coordenadas iniciales características de la especie reaccionante (es decir, dentro de uno de los valles en el lado reactivo de la superficie potencial) y se sigue la trayectoria el tiempo suficiente para determinar si la colisión da como resultado
- un resultado no reactivo caracterizado por coordenadas finales que describen reaccionantes no moléculas de producto, o
- un resultado reactivo que es reconocido por las coordenadas finales que describen las moléculas del producto en lugar de los reactivos.
Se debe hacerlo para un gran número de trayectorias cuyas coordenadas iniciales y momento sean representativos de las condiciones experimentales que se intenta simular. Entonces, hay que promediar los resultados de estas trayectorias sobre este conjunto de condiciones iniciales. En los Capítulos 7 y 8 se discute más sobre cómo se lleva a cabo dicho promedio de conjunto.
Si la molécula contiene más de 3 o 4 átomos, es más común no calcular la energía Born-Oppenheimer en un conjunto de geometrías y luego ajustar estos datos a una forma analítica. En cambio, se inicia una trayectoria en algunas coordenadas iniciales\(\{q_J(0)\}\) y con algunos momentos iniciales\(\{p_J(0)\}\) y luego se utilizan las ecuaciones de Newton, generalmente en la forma de diferencia finita:
\[q_J = q_J(0) + \dfrac{p_J(0)}{m_J} dt \tag{5.3.20}\]
\[p_J = p_J(0) -\dfrac{\partial E}{\partial q_J}(t=0) dt, \tag{5.3.21}\]
para propagar las coordenadas y momenta hacia adelante en el tiempo en una pequeña cantidad\(\delta{t}\). Aquí,\(\dfrac{\partial{E}}{\partial{q_J}}(t=0)\) denota el gradiente de la energía BO calculada a los\(\{q_J(0)\}\) valores de las coordenadas. Luego se vuelve a utilizar el procedimiento de propagación anterior, pero con los valores de\(q_J\) y\(p_J\) apropiados al tiempo\(t = \delta{t}\) como nuevas coordenadas iniciales y momenta, para generar otro conjunto de\(\{q_J\}\) y\(\{p_J\}\) valores. En tales enfoques de dinámica directa, los gradientes de energía, que producen las fuerzas, se calculan solo en geometrías que la trayectoria clásica encuentra a lo largo de su propagación en el tiempo. En el procedimiento anterior, en el que la energía BO se ajusta a una forma analítica, a menudo se calcula\(E\) en geometrías a las que la trayectoria nunca accede.
Al llevar a cabo una simulación de trayectoria tan clásica de una reacción química, hay otras cuestiones que deben abordarse. En particular, como se mencionó anteriormente, esencialmente nunca se puede utilizar ninguna trayectoria única para simular una reacción realizada en un entorno de laboratorio. Uno debe realizar una serie de tales cálculos de trayectoria con una variedad de diferentes coordenadas iniciales y momentos elegidos de manera que representen las condiciones experimentales de interés. Por ejemplo, supongamos que se desea modelar un experimento de haz molecular en el que un haz de especies que\(A\) tiene una energía cinética bien definida\(E_A\) colisiona con un haz de especies\(B\) que tienen energía cinética\(E_B\) como se muestra en la Figura 5.25.
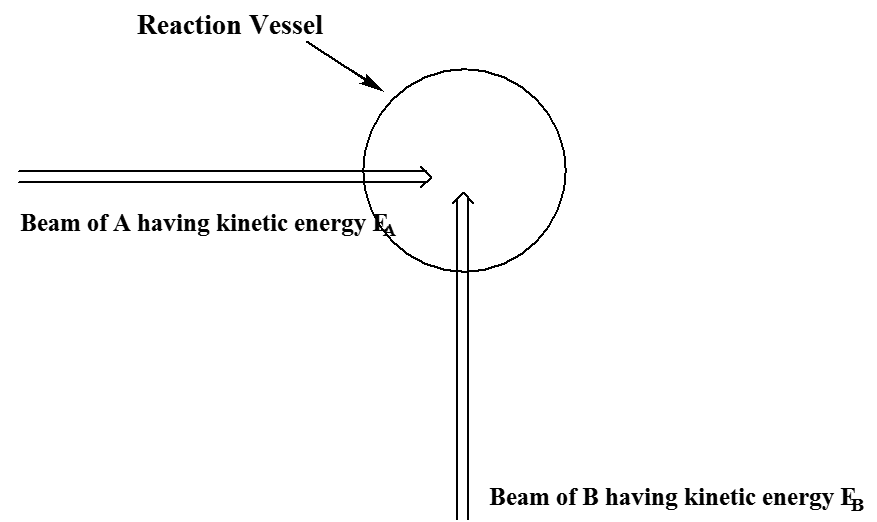
A pesar de que todas\(B\) las moléculas\(A\) y colisionan en ángulos rectos y con energías cinéticas especificadas (y, por lo tanto, momentos iniciales especificados), no todas estas colisiones ocurren de frente. La figura 5.26 ilustra este punto.
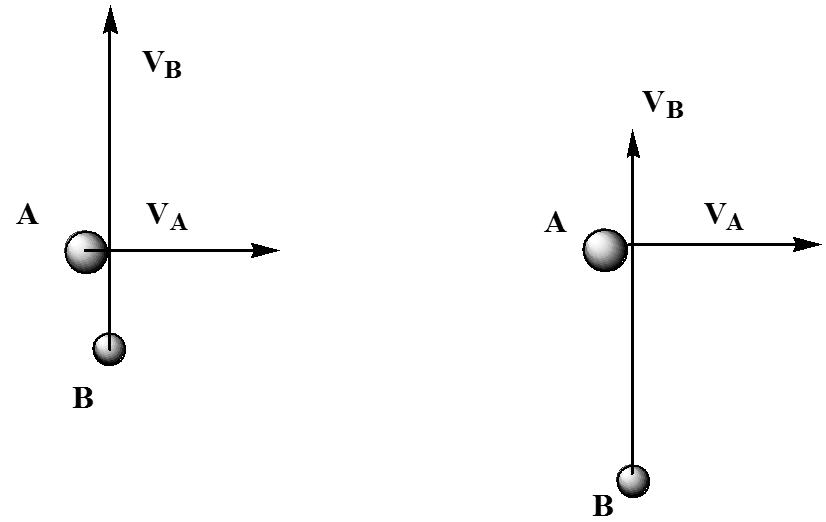
Aquí, mostramos dos colisiones entre una\(A\) y una\(B\) molécula, las cuales tienen idénticas\(A\) y\(B\) velocidades\(V_A\) y\(V_B\), respectivamente. Lo que difiere en los dos eventos es su distancia de aproximación más cercana. En el choque que se muestra a la izquierda, los\(A\) y se\(B\) juntan de cerca. No obstante, en la colisión izquierda, la molécula A se aleja de la región donde la\(B\) golpearía antes de que la\(B\) haya alcanzado. Estos dos casos pueden ser vistos desde una perspectiva diferente que ayuda a aclarar sus diferencias. En la Figura 5.27, se ilustran estas dos colisiones vistas desde un marco de referencia localizado en la\(A\) molécula.
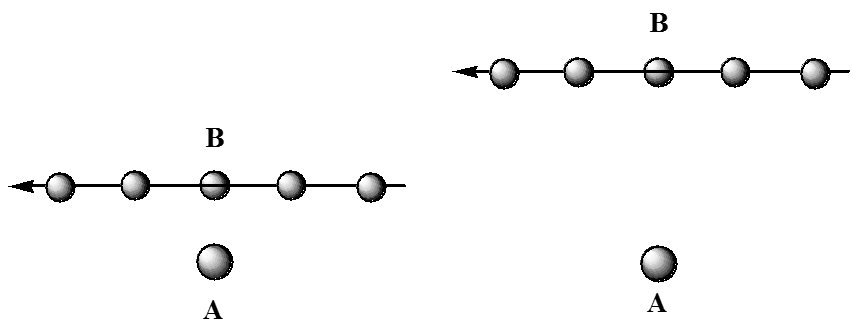
En esta figura, se muestra la ubicación de la\(B\) molécula relativa\(A\) a una serie de veces, mostrando el\(B\) movimiento de derecha a izquierda. En la figura de la izquierda, la\(B\) molécula sufre claramente una colisión más cercana que la de la derecha. La distancia de aproximación más cercana en cada caso se denomina parámetro de impacto y representa la distancia de aproximación más cercana si los socios colisionantes no experimentaron ninguna interacción atractiva o repulsiva (ya que las cifras anteriores serían consistentes con). Por supuesto, cuando\(A\) y\(B\) tengan fuerzas que actúen entre ellas, las trayectorias mostradas anteriormente serían modificadas para parecerse más a las mostradas en la Figura 5.28.
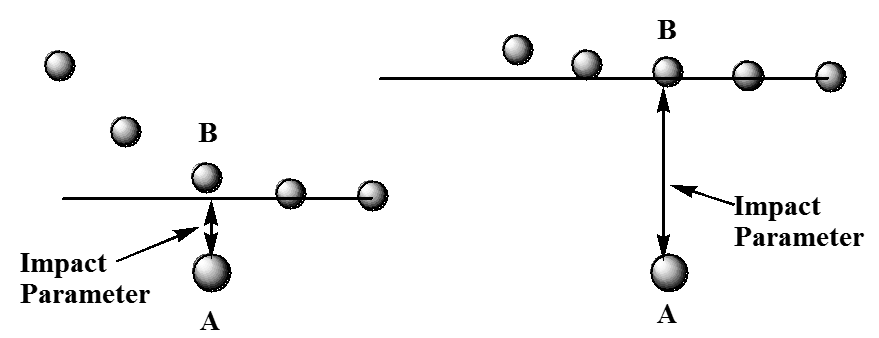
En ambas trayectorias, las fuerzas intermoleculares repulsivas hacen que la trayectoria se aleje de su trayectoria inicial, la cual define los respectivos parámetros de impacto.
Entonces, incluso en este ejemplo de haz molecular en el que ambas moléculas colisionantes tienen velocidades bien especificadas, se deben realizar una serie de trayectorias clásicas, cada una con un parámetro de impacto b diferente para simular el evento de laboratorio. En la práctica, los parámetros de impacto pueden elegirse para variar desde\(b = 0\) (es decir, una cabeza en colisión) hasta algún valor máximo\(b_{Max}\) más allá del cual las\(B\) moléculas\(A\) y ya no interactúan (y por lo tanto ya no pueden sufrir reacción). Cada trayectoria se sigue el tiempo suficiente para determinar si conduce a geometrías características de las moléculas del producto. La fracción de tales trayectorias, ponderada por el elemento de volumen\(2\pi b\,db\) para trayectorias con parámetros de impacto en el rango entre\(b\) y\(b + \delta{b}\), luego da la fracción promediada de trayectorias que reaccionan.
En la mayoría de las simulaciones de reacciones químicas, hay más condiciones iniciales que también deben muestrearse (es decir, se deben seguir trayectorias con una variedad de variables iniciales) y ponderarse adecuadamente. Por ejemplo,
- si existe un rango de velocidades para los reactivos\(A\) y/o\(B\), se deben seguir trayectorias con velocidades en este rango y ponderar los resultados (es decir, reacción o no) de tales trayectorias apropiadamente (por ejemplo, con un factor de ponderación Maxwell-Boltzmann), y
- si las moléculas reaccionantes tienen longitudes de enlace internas, ángulos y orientaciones, se deben seguir trayectorias con diferentes valores iniciales de estas variables y ponderar adecuadamente cada una de tales trayectorias (por ejemplo, usando la distribución de probabilidad de coordenadas del estado vibratorio como factor de ponderación para la inicial valores de esa coordenada).
Como resultado, para simular adecuadamente un experimento de laboratorio de una reacción química, generalmente se requiere que uno siga un número muy grande de trayectorias clásicas. Afortunadamente, tal tarea es muy adecuada para la computación paralela distribuida, por lo que actualmente es factible hacerlo incluso para reacciones bastante complejas.
Existe una situación en la que el enfoque de trayectoria clásica anterior puede ser tonto de perseguir, aunque haya razones para creer que una descripción clásica de Newton de los movimientos nucleares es adecuada. Esto ocurre cuando se tiene una barrera bastante alta para superar para evolucionar de reactivos a productos y cuando la fracción de trayectorias cuyas condiciones iniciales permiten acceder a esta barrera es muy pequeña. En tales casos, uno se enfrenta a que las trayectorias reactivas son muy raras entre el conjunto completo de trayectorias necesarias para simular adecuadamente el experimento de laboratorio. Ciertamente, se puede aplicar la técnica de seguimiento de trayectoria descrita anteriormente, pero si se observa, por ejemplo, que solo una trayectoria en 106 produce una reacción, es posible que no se tengan estadísticas adecuadas para determinar la probabilidad de reacción. Posteriormente se podrían ejecutar 108 trayectorias (elegidas nuevamente para representar el mismo experimento), y ver si 100 o 53 o 212 de estas trayectorias reaccionan, aumentando así la precisión de su probabilidad de reacción. Sin embargo, puede ser computacionalmente poco práctico realizar 100 veces más trayectorias para lograr una mejor precisión en la probabilidad de reacción.
Cuando se enfrentan a tales situaciones de eventos poco frecuentes, generalmente es mejor usar un enfoque que rompa el problema de determinar qué fracción de las condiciones iniciales (debidamente ponderadas) producen reacción en dos partes:
- entre todas las condiciones iniciales (debidamente ponderadas), ¿qué fracción puede acceder a la barrera de alta energía? y
- de los que sí acceden a la barrera alta, ¿cómo puede reaccionar?
Esta forma de formular la pregunta de probabilidad de reacción conduce al método de la teoría del estado de transición (TST) que se trata en detalle en el Capítulo 8, junto con algunas de sus variantes más comunes.
Brevemente, la respuesta a la primera pregunta planteada anteriormente implica calcular la fracción de cuasi-equilibrio de especies reaccionantes que alcanzan la región barrera en términos de las funciones de partición de la mecánica estadística. Este paso se vuelve práctico si se puede suponer que los reactivos químicos están en alguna forma de equilibrio térmico (que es donde son útiles este tipo de modelos). En la forma más simple de TST, la respuesta a la segunda pregunta planteada anteriormente se toma como “reaccionan todas las trayectorias que llegan a la barrera”. En variantes más sofisticadas, se introducen otros modelos para tomar en consideración que no todas las trayectorias que cruzan la barrera efectivamente avanzan hacia los productos y que algunas trayectorias pueden atravesar la barrera cerca de su parte superior. Dejaré más discusión del TST al Capítulo 8.
Además de la trayectoria clásica y los enfoques TST para simular reacciones químicas, hay más enfoques cuánticos. Estas técnicas deben ser utilizadas cuando los núcleos involucrados en la reacción incluyen núcleos de hidrógeno o deuterio. Una discusión de los detalles involucrados en la propagación cuántica está más allá del nivel de este Capítulo, por lo que la retrasaré hasta el Capítulo 8.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)