
8.2: Sondas experimentales de dinámica de reacción
- Page ID
- 70902

Métodos espectroscópicos
Para seguir la velocidad de cualquier reacción química, se debe tener un medio para monitorear las concentraciones de reactivos o moléculas de producto a medida que evoluciona el tiempo. En la mayoría de los experimentos actuales que se relacionan con la dinámica de reacción, se usa alguna forma de sonda física espectroscópica o alternativa (por ejemplo, una firma electroquímica o una detección espectrométrica de masas de iones producto) para monitorear estas concentraciones como funciones del tiempo. Por supuesto, en todas esas mediciones, se debe saber cómo se relaciona la intensidad de la señal detectada con la concentración de las moléculas que causan la señal. Por ejemplo, en muchos experimentos de absorción, como se ilustra en la Figura 8.4, la luz se pasa a través de una muestra de espesor\(L\) y se mide la intensidad del haz de luz en ausencia de la muestra\(I_0\) y con la muestra presente\(I\).
La ley Beer-Lambert:
\[\log(I_0/I) = \varepsilon[A]L\]
entonces permite determinar la concentración [A] de las moléculas absorbentes, dada la longitud de la trayectoria\(L\) sobre la que ocurre la absorción y dado el coeficiente\(\varepsilon\) de extinción de las moléculas absorbentes.
Estos coeficientes de extinción, que se relacionan con los elementos de la matriz dipolo eléctrico como se discute en el Capítulo 6, generalmente se determinan empíricamente preparando una concentración conocida de las moléculas absorbentes y midiendo la\(I_0/I\) relación que esta concentración produce en una célula de longitud\(L\). Para moléculas e iones que son extremadamente reactivos, este enfoque de calibración para determinar a menudo no\(\varepsilon\) es factible porque no se puede preparar una muestra con una concentración conocida que permanece constante en el tiempo el tiempo suficiente para que se lleve a cabo el experimento. En tales casos, a menudo se debe recurrir al uso de las expresiones teóricas dadas en el Capítulo 6 (y discutidas en la mayoría de los libros de texto sobre espectroscopia molecular) para calcular\(\varepsilon\) en términos de las funciones de onda de la especie absorbente. En cualquier caso, se debe saber cómo se relaciona la intensidad de la señal con las concentraciones de la especie si se desea monitorear la reacción química o las velocidades de transferencia de energía.
Debido a que las técnicas experimentales modernas son capaces de detectar moléculas en particular estados electrónicos y vibración-rotación, se ha vuelto común utilizar tales herramientas para examinar la dinámica de reacción química a nivel de estado a estado y para seguir procesos de transferencia de energía, que claramente requieren tal estado- datos específicos. En tales experimentos, se busca conocer la velocidad a la que\(\Phi_i\) reaccionan los reactivos en un estado específico para producir productos en algún estado específico\(\Phi_f\). Una de las formas más comunes de monitorear tales tasas específicas de estado es a través de un experimento denominado bomba-sonda en el que
i. Se utiliza un pulso de luz de corta duración para excitar moléculas reaccionantes a algún estado inicial especificado\(\Phi_i\). Por lo general, se usa un láser sintonizable porque su dispersión de frecuencia estrecha permite bombear estados específicos. El tiempo en el que este láser de bombeo prepara así las moléculas reaccionantes excitadas en estado\(\Phi_i\) define\(t = 0\).
ii. Después de un tiempo de retardo de duración t, se utiliza una segunda fuente de luz para sondear las moléculas de producto que se han formado en diversos estados finales,\(\Phi_f\). A menudo, se escanea la frecuencia de esta fuente de sonda para que se puedan examinar poblaciones de muchos de estos estados finales.
Las concentraciones de reactivos y moléculas de productos en los estados inicial y final\(\Phi_i\) y\(\Phi_f\) están determinadas por la relación Beer-Lambert asumiendo que se conocen los coeficientes de extinción\(\varepsilon_i\) y\(\varepsilon_f\) para estas especies y estados de absorción. En el primer caso, el coeficiente de extinción\(\varepsilon_i\) se relaciona con la absorción de los fotones de la bomba para preparar moléculas reaccionantes en el estado inicial especificado. En este último,\(\varepsilon_f\) se refiere a la absorción de las moléculas del producto que se crean en el estado\(\Phi_f\). Llevar a cabo una serie de tales mediciones de absorción de estado final en diversos tiempos de retardo t permite determinar la concentración de estos estados en función del tiempo.
Este tipo de experimento de bomba-sonda láser se utiliza no solo para sondear estados electrónicos o de vibración/rotación específicos de los reactivos y productos, sino también cuando la reacción es rápida (es decir, completa en 10 -4 s o menos). En estos casos, no se está utilizando la resolución de alta frecuencia del láser sino su rápida respuesta de tiempo. Debido a que se pueden generar pulsos láser de duración bastante corta, estas herramientas son muy adecuadas en estudios de reacciones químicas tan rápidas. Las reacciones pueden ser en fase gaseosa (por ejemplo, reacciones radicales rápidas en la atmósfera o en explosiones) o en solución (por ejemplo, reacciones de transferencia de electrones fotoinducidas en sistemas biológicos).
Métodos de haz
Otro enfoque para sondear la dinámica de reacción química es usar un haz de moléculas reaccionantes A que colisionen con otros reactivos B que también pueden estar en un haz o en un bulbo en equilibrio a alguna temperatura T. Dichos experimentos de haz cruzado y bulbo de haz se ilustran en la Figura 8.5.
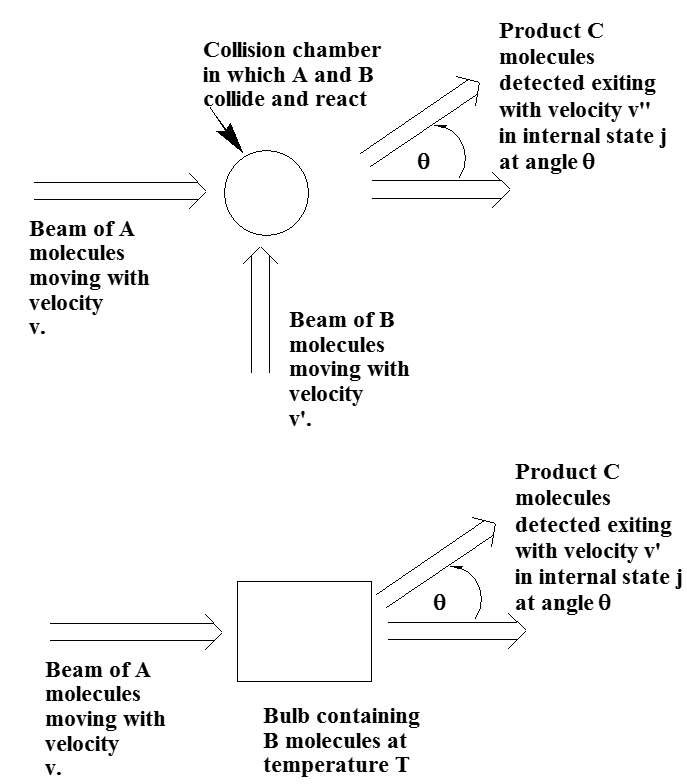
Casi siempre, estas muestras de haz y bulbo contienen moléculas, radicales o iones en fase gaseosa, por lo que estas técnicas son más prevalentes en estudios de dinámica en fase gaseosa.
Las ventajas de los experimentos de tipo viga cruzada son que:
- se pueden controlar las velocidades, y por lo tanto las energías de colisión, de ambos reactivos,
- se puede examinar el rendimiento del producto en función del ángulo\(\theta\) a través del cual se dispersan los productos,
- se puede sondear la velocidad de los productos y,
- mediante el uso de métodos espectroscópicos, se puede determinar la fracción de productos generados en diversos estados internos (electrónico/vibracional/rotacional).
Dichas mediciones permiten obtener información muy detallada sobre cómo el coeficiente de velocidad de reacción depende de la energía colisional (cinética) y dónde se deposita la energía total disponible para los productos (es decir, en energía traslacional del producto o energía interna del producto). La distribución angular de las moléculas del producto también puede dar información sobre la naturaleza del proceso de reacción. Por ejemplo, si la colisión A + B forma un complejo de colisión de larga duración (es decir, en escalas de tiempo de rotación), las moléculas C del producto muestran una distribución angular muy isotrópica. Por el contrario, las reacciones que proceden de manera más impulsiva muestran distribuciones angulares del producto que están fuertemente retrodispersadas o fuertemente dispersadas hacia adelante en lugar de isotrópicas.
En experimentos de haz y bulbo, uno no es capaz de obtener tanta información detallada porque una de las moléculas reaccionantes B no está obligada a moverse con una velocidad fija conocida en una dirección especificada cuando ocurren las\(A + B \rightarrow C\) colisiones. En cambio, las moléculas B chocan con moléculas A en una variedad de orientaciones y con una distribución de energías de colisión cuyo rango depende de la distribución Maxwell-Boltzmann de las energías cinéticas de las moléculas B en el bulbo. La ventaja de los experimentos de haz y bulbo es que se pueden lograr densidades de colisión mucho más altas que en los experimentos de haz cruzado porque la densidad de las moléculas B dentro del bulbo puede ser mucho mayor que las densidades alcanzables en un haz de moléculas B.
Hay casos en los que los experimentos de bulbo de haz pueden ser utilizados para determinar cómo la velocidad de reacción depende de la energía de colisión a pesar de que las moléculas en el bulbo tienen una distribución de energías cinéticas. Es decir, si las especies en el haz tienen energías cinéticas mucho más altas que la mayoría de las moléculas B, entonces la energía de colisión A + B está determinada principalmente por la energía del haz. Un ejemplo de esta situación lo proporcionan los denominados experimentos de haz de iones guiados en los que un haz de iones que tiene una energía cinética E bien especificada incide sobre moléculas en una bombilla que tiene una temperatura\(T\) para la cual\(kT \ll E\). La Figura 8.6 ilustra datos que pueden extraerse de dicho experimento.
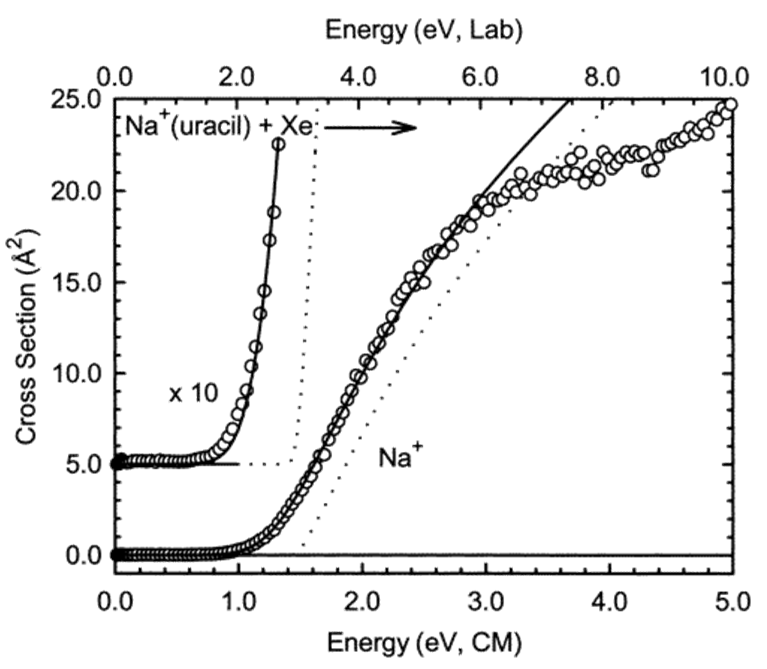
En la Figura 8.6, se ilustra la sección transversal\(\sigma\) (relacionada con la constante de velocidad bimolecular\(k\) por\(\sigma v = k\), donde v es la velocidad de colisión relativa) para la producción de\(Na^+\) iones cuando un haz de complejos\(Na^+\) (uracilo) que tienen energía E (el eje horizontal) colisiona con una bombilla conteniendo átomos de Xe a temperatura ambiente. En este caso, la reacción es simplemente el proceso de disociación inducida por colisión (CID) en el que el complejo experimenta descomposición unimolecular después de obtener energía interna en colisiones con átomos de Xe:
\[Na^+({\rm uracil}) \rightarrow Na^+ + \rm uracil.\]
El conocimiento principal adquirido en este experimento CID es la energía umbral\(E^*\); es decir, la energía mínima de colisión necesaria para efectuar la disociación del\(Na^+({\rm uracil})\) complejo. Este tipo de datos han demostrado ofrecer algunos de los datos más útiles sobre las energías de disociación de enlaces de una amplia variedad de especies. Además, la magnitud de la sección transversal\(\sigma\) de la reacción en función de la energía de colisión es un producto valioso de tales experimentos. Este tipo de experimentos CID con bulbo de haz ofrecen uno de los medios más potentes y ampliamente utilizados para determinar tales energías de ruptura de enlaces y constantes de velocidad de reacción.
Otros métodos
Por supuesto, no todas las reacciones químicas ocurren tan rápidamente que requieren el uso de láseres rápidos para seguir concentraciones de especies reaccionantes o técnicas de bomba-sonda para generar y sondear estas moléculas. Para reacciones químicas más lentas, se pueden utilizar otros métodos para monitorear las concentraciones relevantes. Estos métodos incluyen la electroquímica (donde el potencial redox es la firma de la especie) y la espectroscopia de RMN (donde los desplazamientos químicos de los grupos funcionales son las firmas) cuyos tiempos de respuesta instrumental son demasiado lentos para sondear reacciones rápidas.
Además, cuando las reacciones en estudio no proceden a su finalización sino que existen en equilibrio con una reacción posterior, se pueden utilizar enfoques alternativos. El ejemplo que se discute en el Capítulo 5 es uno de esos casos. Revisemos brevemente aquí y nuevamente consideremos la reacción de una enzima E y un sustrato S para formar el complejo enzima-sustrato ES:
\[E + S \rightleftharpoons ES.\]
En los experimentos de tipo perturbación, las concentraciones de equilibrio de la especie se “desplazan” en una pequeña cantidad\(\delta\) por aplicación de la perturbación, de modo que
\[[ES] = [ES]_{\rm eq} -\delta\]
\[[E] = [E]_{\rm eq} +\delta\]
\[[S] = [S]_{\rm eq} +\delta.\]
Posteriormente, la siguiente ley de tasas regirá la evolución temporal del cambio de concentración d:
\[-\dfrac{d\delta}{dt} = - k_r ([ES]_{\rm eq} -\delta) + k_f ([E]_{\rm eq} +\delta) ([S]_{\rm eq} +\delta).\]
Suponiendo que eso\(\delta\) es muy pequeño (para que se descuide el término que implica\(\delta^2\) cam) y utilizando el hecho de que las tasas hacia adelante y hacia atrás se equilibran en equilibrio, esta ecuación para la evolución temporal de\(\delta\) puede reducirse a:
\[-\dfrac{d\delta}{dt} = (k_r + k_f [S]_{\rm eq} + k_f[E]_{\rm eq}) \delta.\]
Entonces, las desviaciones de concentración del equilibrio volverán al equilibrio exponencialmente con un coeficiente de tasa efectivo que es igual a una suma de términos:
\[k_{\rm eff} = k_r + k_f [S]_{\rm eq} + k_f[E]_{\rm eq}.\]
Entonces, siguiendo las concentraciones de los reactivos o productos a medida que regresan a sus valores de equilibrio, se puede extraer el coeficiente de tasa efectiva\(k_{\rm eff}\). Haciendo esto a una variedad de diferentes concentraciones iniciales de equilibrio (e, g.,\([S]_{\rm eq}\) y\([E]_{\rm eq}\)), y viendo cómo\(k_{\rm eff}\) cambia, se pueden determinar las constantes de velocidad tanto hacia adelante como hacia atrás.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)