
9: Ejercicios
- Page ID
- 71042

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A continuación se presentan algunos problemas que te ayudarán a refrescar tu memoria sobre el material que debiste haber aprendido en las clases de química de pregrado y que te permitirán ejercitar el material enseñado en este texto.
Capítulos de la Parte 1
1.Usted debe ser capaz de configurar y resolver la partícula unidimensional y bidimensional en una caja ecuaciones de Schrödinger. Te sugiero que pruebes esto y asegúrate de ver:
- Cómo las ecuaciones diferenciales de segundo orden tienen dos soluciones independientes, por lo que la solución más general es una suma de estas dos.
- Cómo las dos condiciones límite reducen el número de soluciones aceptables de dos a uno y limitan los valores de\(E\) que se pueden “permitir”.
- Cómo la función de onda es continua incluso en los límites de la caja, pero no lo\(\dfrac{d\Psi}{dx}\) es. En general\(\dfrac{d}{dx}\), lo que se relaciona con el impulso porque\(-i\hbar \dfrac{d}{dx}\) es el operador de impulso, es continuo excepto en puntos donde el potencial\(V(x)\) sufre un salto infinito como lo hace en los límites de la caja. El salto infinito\(V\), cuando se ve clásicamente, significa que la partícula sufriría una inversión instantánea de impulso en este punto, por lo que su impulso no sería continuo. Por supuesto, en cualquier sistema realista,\(V\) no tiene saltos infinitos, por lo que el impulso variará sin problemas y así\(\dfrac{d\Psi}{dx}\) será continuo.
- Cómo crecen los niveles de energía con el número cuántico\(n\) como\(n^2\).
- Cómo se ven las funciones de onda cuando se trazan.
2. Debe pasar por las diversas funciones de onda tratadas en la Parte 1 (por ejemplo, partículas en cajas, rotor rígido, oscilador armónico) y asegurarse de ver cómo las gráficas de\(|\Psi|^2\) probabilidad de tales funciones no son en absoluto como las distribuciones de probabilidad clásicas excepto cuando el número cuántico es muy grande .
3. Debes asegurarte de entender cómo la evolución temporal de un autoestado\(\Psi\) produce un simple\(\exp(-i tE/ \hbar)\) múltiplo de\(\Psi\) para que\(|\Psi|^2\) no dependa del tiempo. Sin embargo, cuando no\(\Psi\) es un estado propio (por ejemplo, cuando es una combinación de tales estados), su propagación temporal produce una\(\Psi\) cuya distribución de\(|\Psi|^2\) probabilidad cambia con el tiempo.
4. Debe notar que las densidades de estados apropiados a la partícula 1-, 2- y 3-dimensional en un problema de caja (que se relacionan con las traducciones en estas dimensiones) dependen de diferentes potencias de\(E\) para las diferentes dimensiones.
5. Deberías poder resolver problemas de valores propios de la matriz de Hückel 2x2 y 3x3 tanto para obtener las energías orbitales como los vectores propios normalizados. Para la práctica, tratar de hacerlo para
- los tres\(\pi\) orbitales del radical alilo
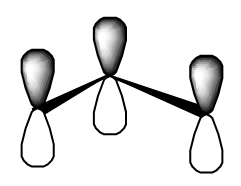
- los tres\(\pi\) orbitales del radical ciclopropenly.
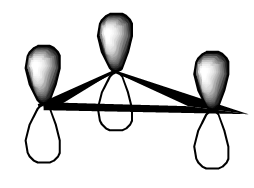
¿Ves que el álgebra necesario para encontrar los conjuntos de orbitales anteriores es exactamente el mismo que se necesitaba cuando tratamos el trímero de sodio lineal y triangular?
6. Debe ser capaz de seguir la derivación de la probabilidad de tunelización. Hacer esto ofrece una buena prueba de tu capacidad para aplicar las condiciones de contorno correctamente, por lo que te sugiero que hagas esta tarea. Debe apreciar cómo la probabilidad de tunelización decae exponencialmente con el “grosor” de la barrera de tunelización y con la “altura” de esta barrera y que la tunelización para partículas más pesadas es menos probable que para partículas ligeras. Esta es la razón por la que la tunelización generalmente se considera solo para electrones, protones y neutrones.
7. No espero que se pueda llevar una solución completa a la ecuación de Schrödinger para el átomo hidrogénico. No obstante, creo que hay que prestar atención a
- Cómo las separaciones de variables conducen a una radial y dos ecuaciones diferenciales angulares de segundo orden.
- Cómo la condición límite que y + 2 son puntos equivalentes en el espacio produce el número cuántico m.
- Cómo surge el número cuántico l de la ecuación.
- Cómo la condición de que la función de onda radial no “explote” (es decir, ir al infinito) a medida que la coordenada r se vuelve grande da lugar a la ecuación para la energía\(E\).
- El hecho de que las partes angulares de las funciones de onda sean armónicos esféricos, y que éstas sean exactamente las mismas funciones de onda para el movimiento rotacional de una molécula lineal.
- Cómo\(E\) depende la energía del número\(n\) cuántico como\(n^{-2}\) y de la carga nuclear\(Z\) como\(Z^2\), y que las energías del estado unido son negativas (¿entiendes lo que esto significa? Es decir, ¿cuál es el cero o punto de referencia de la energía?).
8. Debes asegurarte de estar familiarizado con cómo las energías rígido-rotor y oscilador armónico varían con los números cuánticos (\(J\),\(M\) en el primer caso,\(v\) en el segundo). También debes saber cómo estas energías dependen de la geometría molecular (en la primera) y de la fuerza constante y de la masa reducida (en la segunda). Debe tener en cuenta que\(E\) depende cuadráticamente de\(J\) pero linealmente de\(v\).
9. Debes saber qué es el potencial Morse y qué significan sus parámetros. Se debe entender que el potencial Morse muestra anarmonicidad, pero el potencial armónico no.
10. Deberías poder seguir cómo se puede usar la matriz de Hessian ponderada en masa para aproximar los movimientos vibracionales de una molécula poliatómica. Y, se debe entender cómo los valores propios de esta matriz producen las frecuencias vibracionales armónicas y los vectores propios correspondientes describen los movimientos de la molécula asociados a estas frecuencias.
Practicar con matrices y operadores
1.Encuentre los valores propios y los vectores propios normalizados correspondientes de las siguientes matrices:
\ [\ left [\ begin {array} {cc}
-1 & 2\\
2 & 2
\ end {array}\ derecha]\]
\ [\ left [\ begin {array} {ccc}
-2 & 0 & 0\\
0 & -1 & 2\\
0 & 2 & 2 & 2
\ end {array}\ derecha]\]
2. Reemplazar las siguientes expresiones mecánicas clásicas por sus operadores mecánicos cuánticos correspondientes:
- K.E. =\(\dfrac{mv^2}{2}\) en el espacio tridimensional.
- \(\textbf{p} = m\textbf{v}\), un vector cartesiano tridimensional.
- \(y\)-componente de momento angular:\(L_y = zp_x - xp_z\).
3. Transforme los siguientes operadores en las coordenadas especificadas:
- \(L_x=\)de coordenadas polares cartesianas a esféricas.
- \(L_z=\)desde coordenadas polares esféricas a cartesianas.
4. Hacer coincidir las funciones propias de la columna B con sus operadores en la columna A. ¿Cuál es el valor propio para cada función propia?
\ [\ begin {array} {lll}
\ phantom {aaaaa} &\ text {Columna A} &\ texto {Columna B}\\\ hline
{\ rm i.} & (1-x^2) - x & 4x^4 - 12x^2 + 3\\
{\ rm ii.} &\ dfrac {d^2} {dx^2} y 5x^4\\
{\ rm iii.} & x\ dfrac {d} {dx} & e^ {3x} + e^ {-3x}\\
{\ rm iv.} &\ dfrac {d^2} {dx^2} - 2x\ dfrac {d} {dx} & x^2 - 4x + 2\\
{\ rm v.} & x\ dfrac {d^2} {dx^2} + (1-x)\ dfrac {d} {dx} & 4x^3 - 3x
\ end {array}\]
Revisión de formas de orbitales
5.Dibujar formas cualitativas de los orbitales\(d\) atómicos (1)\(s\), (3)\(p\) y (5) (tenga en cuenta que estos orbitales representan solo la porción angular y no contienen la porción radial del hidrógeno como las funciones de onda atómica) Indicar con los signos relativos de las funciones de onda y el posición (s) (si la hay) de cualquier nodo.
6.Trazar las porciones radiales de las\(4s\) funciones de onda atómica\(4d\), e\(4f\) hidrógeno.\(4p\)
7. Trazar las porciones radiales de las\(1s\) funciones de onda atómica similares a\(3p\) hidrógeno para el átomo de Si usando conceptos de cribado para cualquier electrón interno.\(2s\)\(2p\)\(3s\)
Etiquetado de orbitales usando simetría de grupo de puntos
8. Definir los orbitales atómicos “núcleo” y “valencia” adaptados a la simetría de los siguientes sistemas:
- \(NH_3\)en el grupo\(C_{3v}\) de puntos,
- \(H_2O\)en el grupo\(C_{2v}\) de puntos,
- \(H_2O_2\)(cis) en el grupo\(C_2\) de puntos
- \(N\)en\(D_{\infty h}\),\(D_{2h}\)\(C_{2v}\), y grupos\(C_{s}\) puntuales
- \(N_2\)en\(D_{\infty h}\),\(D_{2h}\)\(C_{2v}\), y grupos\(C_{s}\) puntuales.
problema para practicar las herramientas básicas de la ecuación de Schrödinger
9. Una partícula de masa\(m\) se mueve en una caja unidimensional de longitud\(L\), con límites en\(x = 0\) y\(x = L\). Así,\(V(x)\) = 0 para\(0 \le x \le L\), y\(V(x) = \infty\) en otros lugares. Las funciones propias normalizadas del hamiltoniano para este sistema están dadas por\(\Psi_n(x) = \sqrt{\dfrac{2}{L}} \sin\dfrac{2\pi x}{L}\), con\(E_n =\dfrac{n^2\pi^2\hbar^2}{2mL^2}\), donde el número cuántico\(n\) puede tomar los valores\(n=1,2,3,....\)
- Suponiendo que la partícula está en un estado propio,\(\Psi_n(x)\), calcular la probabilidad de que la partícula se encuentre en algún lugar de la región\(0 \le x \le \dfrac{L}{4}\). Mostrar de qué depende esta probabilidad\(n\).
- ¿Para qué valor de\(n\) existe la mayor probabilidad de encontrar la partícula en\(0 \le x \le \dfrac{L}{4}\)?
- Ahora supongamos que\(\Psi\) es una superposición de dos autoestados,\[\Psi = a\Psi_n + b\Psi_m, \text{ at time } t = 0. \] ¿Qué es\(\Psi\) en el tiempo t? ¿Qué valor de expectativa energética\(\Psi\) tiene en el tiempo t y cómo se relaciona esto con su valor en\(t = 0\)?
- Para una medición experimental que sea capaz de distinguir sistemas en estado\(\Psi_n\) de aquellos en\(\Psi_m\), ¿qué fracción de un gran número de sistemas descritos por cada uno se\(\Psi\) observará en estar\(\Psi_n\)? ¿Qué energías encontrarán estas mediciones experimentales y con qué probabilidades?
- Para aquellos sistemas originalmente en los\(\Psi = a\Psi_n + b\Psi_m\) que se observó que estaban en el\(\Psi_n\) momento\(t\), ¿en qué estado (\(\Psi_n\)\(\Psi_m\),, o lo que sea) se encontrarán si se realiza una segunda medición experimental en un momento\(t'\) posterior al\(t\)?
- Supongamos que por algún método (que no necesita preocuparnos en este momento) el sistema ha sido preparado en un estado no estacionario (es decir, no es una función propia de\(H\)). En el momento de una medición de la energía de la partícula, este estado es especificado por la función de onda normalizada\(\Psi = \sqrt{\dfrac{30}{L^5}}x(L-x)\) para\(0 \le x \le L\), y\(\Psi = 0\) en otros lugares. ¿Cuál es la probabilidad de que una medición de la energía de la partícula dé el valor\(E_n =\dfrac{n^2\pi^2\hbar^2}{2mL^2}\) para cualquier valor dado de\(n\)?
- ¿Cuál es el valor esperado de\(H\), es decir, la energía promedio del sistema, para la función de onda\(\Psi\) dada en la parte f?
problema sobre las propiedades de los estados no estacionarios
10. Demostrar que para un sistema en un estado no estacionario,
\(\Psi = \sum_j C_j\Psi_j e^{-iE_jt/\hbar}\), el valor promedio de la energía no varía con el tiempo pero los valores de expectativa de otras propiedades sí varían con el tiempo.
problema sobre la distorsión de Jahn-Teller
11. Los estados de energía y las funciones de onda para una partícula en una caja tridimensional cuyas longitudes son\(L_1\)\(L_2\), y\(L_3\) están dadas por
\[E(n_1,n_2,n_3) = \dfrac{h^2}{8m}\left[\Big(\dfrac{n_1}{L_1}\Big)^2+\Big(\dfrac{n_2}{L_2}\Big)^2+\Big(\dfrac{n_3}{L_3}\Big)^2\right]\text{ and}\]
\[\Psi(n_1,n_2,n_3) = \sqrt{\dfrac{2}{L_1}}\sqrt{\dfrac{2}{L_2}}\sqrt{\dfrac{2}{L_3}} \sin\Big(\dfrac{n_1\pi x}{L_1}\Big) \sin\Big(\dfrac{n_2\pi x}{L_2}\Big) \sin\Big(\dfrac{n_3\pi x}{L_3}\Big).\]
Estas funciones de onda y niveles de energía a veces se utilizan para modelar el movimiento de los electrones en un átomo metálico central (o ion) que está rodeado por seis ligandos de manera octaédrica.
- Mostrar que el nivel de energía más bajo es no degenerado y el segundo nivel de energía es triplicamente degenerado si\(L_1= L_2= L_3\). ¿Qué valores de\(n_1\)\(n_2\), y\(n_3\) caracterizan a los estados pertenecientes al nivel triple degenerado?
- Para una caja de volumen\(V = L_1L_2L_3\) muestran que para tres electrones en la caja (dos en el “orbital” más bajo no degenerado, y uno en el siguiente), se obtendrá una energía total menor si la caja sufre una distorsión rectangular (\(L_1= L_2\ne L_3\)). que conserva el volumen total que si la caja permanece sin distorsionar (indicio: si \(V\)es fijo y\(L_1 = L_2\), entonces\(L_3 =\dfrac{V}{L_1^2}\) y\(L_1\) es la única “variable”).
- Demostrar que el grado de distorsión (relación de\(L_3\) a\(L_1\)) que minimizará la energía total es\(L_3 = \sqrt{2}L_1\). ¿Cómo se relaciona este problema con las distorsiones de Jahn-Teller? ¿Por qué (en términos de la propiedad del átomo o ion central) hacemos el cálculo con volumen fijo?
- Por cuanto (en eV) la distorsión bajará la energía (de su valor para un cubo,\(L_1= L_2= L_3\)) si\(V\) = 8 Å y = 6.01 x 10 erg cm 2. 1 eV = 1.6 x 10 erg
partícula en un modelo de anillo para electrones que se mueven en compuestos cíclicos
12. \(\pi\)Los orbitales del benceno,\(C_6H_6\), pueden modelarse de manera muy crudamente usando las funciones de onda y las energías de una partícula en un anillo. Primero vamos a tratar la partícula en un problema de anillo y luego extenderla al sistema de benceno.
- Supongamos que una partícula de masa m está restringida a moverse en un círculo (de radio\(r\)) en el\(xy\) plano. Asumir además que la energía potencial de la partícula es constante (elija cero como este valor). Anote la ecuación de Schrödinger en la representación de coordenadas cartesianas normales. Transformar esta ecuación de Schrödinger en coordenadas cilíndricas donde\(x = r\cos\theta\)\(y = r\sin\phi\), y\(z = z\) (\(z = 0\)en este caso). Tomando\(r\) para mantenerse constante, anote la solución general,\(\Phi(\phi)\), a esta ecuación de Schrödinger. Las condiciones de “límite” para este problema lo requieren\(\Phi(\phi) = \Phi(\phi+2\pi)\). Aplicar esta condición de límite a la solución general. Esto da como resultado la cuantificación de los niveles de energía de este sistema. Anote la expresión final para las funciones de onda normalizadas y las energías cuantificadas. ¿Cuál es la significación física de estos números cuánticos que pueden tener valores tanto positivos como negativos? Dibuja un diagrama de energía que represente los cinco primeros niveles de energía.
- Tratar los seis\(\pi\) electrones del benceno como partículas libres para moverse en un anillo de radio 1.40 Å, y calcular la energía de la transición electrónica más baja. ¡Asegúrate de que el principio Pauli esté satisfecho! ¿A qué longitud de onda corresponde esta transición? Sugieran algunas razones por las que esto difiere de la longitud de onda de la transición más baja observada en benceno, que es de 2600 Å.
función de onda de estado no estacionario
13. Una molécula diatómica obligada a girar sobre una superficie plana puede modelarse como un rotor rígido plano (con funciones propias\(\Phi(\phi)\), análogas a las de la partícula en un anillo del problema 12) con longitud de enlace fija\(r\). At\(t = 0\), la distribución de probabilidad rotacional (orientacional) se observa para ser descrita por una función de onda\(\Psi(\phi,0) = \sqrt{\dfrac{4}{3\pi}}\cos^2\phi\). ¿Qué valores, y con qué probabilidades, del momento angular rotacional\(-i\hbar\dfrac{\partial}{\partial \phi}\), podrían observarse en este sistema? Explique si estas probabilidades serían dependientes del tiempo a medida que\(\Psi(\phi,0)\) evoluciona hacia\(\Psi(\phi,t)\).
problema sobre los factores Franck-Condon
14. Consideremos una\(N_2\) molécula, en el nivel vibratorio terrestre del estado electrónico de tierra, que es bombardeada por electrones de 100 eV. Esto lleva a la ionización de la\(N_2\) molécula para que se forme\(N\). En este problema intentaremos calcular la distribución vibratoria de los\(N\) iones recién formados, utilizando un enfoque algo simplificado.
- Calcular (según la mecánica clásica) la velocidad (en cm/s) de un electrón de 100 eV, ignorando cualquier efecto relativista. También calcule la cantidad de tiempo requerido para que un electrón de 100 eV pase una Nmolécula, que puede estimar que tiene una longitud de 2Å.
- La ecuación radial de Schrödinger para una molécula diatómica que trata la vibración como un oscilador armónico puede escribirse como:
\[ -\dfrac{\hbar^2}{2\mu r^2}\left(\dfrac{\partial}{\partial r}\left( r^2 \dfrac{\partial \Psi}{\partial r}\right)\right)+ \dfrac{k}{2}(r-r_e)^2 \Psi =E\Psi,\]
Sustituyendo\(\Psi(r) =\dfrac{F(r)}{r} \), esta ecuación se puede reescribir como:
\[-\dfrac{\hbar^2}{2\mu r^2}\dfrac{\partial^2}{\partial r^2}F(r) + \dfrac{k}{2}(r-r_e)^2F(r) = EF(r) .\]
El hamiltoniano vibracional para el estado electrónico básico de la\(N_2\) molécula dentro de esta aproximación viene dado por:
\[H({\rm N_2}) = \dfrac{\hbar^2}{2\mu r^2}\dfrac{\partial^2}{\partial r^2}+ \dfrac{k_{\rm N_2}}{2}(r-r_{\rm N_2})^2,\]
donde\(r_{\rm N_2}\) y se\(k_{\rm N_2}\) hayan medido experimentalmente para ser:
\[ r_{\rm N_2} = 1.09769 Å; \hspace{1cm} k_{\rm N_2} = 2.294 \times 10^6 \dfrac{\rm g}{\rm sec^2}.\]
El hamiltoniano vibracional para el\(N_2^+\) ion, sin embargo, viene dado por:
\[H({\rm N_2^+}) = \dfrac{\hbar^2}{2\mu r^2}\dfrac{\partial^2}{\partial r^2}+ \dfrac{k_{\rm N_2^+}}{2}(r-r_{\rm N_2^+})^2,\]
donde\(r_{\rm N_2^+}\) y se\(k_{\rm N_2^+}\) hayan medido experimentalmente para ser:
\[r_{\rm N_2^+}= 1.11642 Å;\hspace{1cm} k_{\rm N_2} = 2.009 \times 10^6 \dfrac{\rm g}{\rm sec^2}.\]
En ambos sistemas la masa reducida es\(\mu = 1.1624 \times 10^{-23}\) g. Utilice la información anterior para escribir las funciones de onda vibratoria del estado fundamental de las\(N\) moléculas\(N_2\) y, dando valores explícitos para cualquier constante que aparezca en ellas. La función del oscilador\(v = 0\) armónico es\(\Psi_0 = \Big(\dfrac{\alpha}{\pi}\Big)^{1/4} \exp(-\alpha x^2/2)\).
c. Durante la escala de tiempo del evento de ionización (que usted calculó en la parte a), la función de onda vibratoria de la\(N_2\) molécula efectivamente no tiene tiempo para cambiar. Como resultado, el\(N\) ion recién formado se encuentra en un estado vibracional que no es una función propia del nuevo hamiltoniano vibracional,\(H({\rm N}_2^+)\). Suponiendo que la\(N_2\) molécula estaba originalmente en su estado\(v=0\) vibracional, calcular la probabilidad de que el\(N\) ion se produzca en su estado\(v=0\) vibracional.
Vibración de una molécula diatómica
15.La constante de fuerza,\(k\), del\(C-O\) enlace en monóxido de carbono es de 1.87 x 10 g/seg. Supongamos que el movimiento vibracional de\(CO\) es puramente armónico y usa la masa reducida\(\mu= 6.857\) amu.
Calcular el espaciamiento entre los niveles de energía vibratoria en esta molécula, en unidades de ergs y cm -1.
Calcular la incertidumbre en la distancia internuclear en esta molécula, asumiendo que se encuentra en su nivel vibratorio terrestre. Utilice la función de onda vibratoria del estado fundamental (\(\Psi_{v=0}\); recuerde que le di esta función en el problema 14), y calcule\(\langle x\rangle \),\(\langle x^2\rangle \), y\(\Delta x = \sqrt{\langle x^2\rangle - \langle x\rangle ^2}\).
¿Bajo qué circunstancias (es decir, valores grandes o pequeños de\(k\); valores grandes o pequeños de\(\mu\)) es grande la incertidumbre en la distancia internuclear? ¿Se te ocurre alguna relación entre esta observación y el hecho de que el helio sigue siendo un líquido hasta el cero absoluto?
Problema del método variacional
16. Una partícula de masa m se mueve en un potencial unidimensional cuyo hamiltoniano viene dado por
\[H = -\dfrac{\hbar^2}{2m}\dfrac{d^2}{dx^2}+ a|x| ,\]
donde la función de valor absoluto se define por\(|x| = x\) if\(x \ge 0\) y\(|x| = -x\) if\(x \le 0\).
- Utilice la función de onda de prueba normalizada\(\phi = \Big(\dfrac{2b}{\pi}\Big)^{\frac{1}{4}}e^{-bx^2}\) para estimar la energía del estado fundamental de este sistema, utilizando el principio variacional para evaluar\(W(b)\), el valor de expectativa variacional de\(H\).
- Optimizar b para obtener la mejor aproximación a la energía del estado fundamental de este sistema, utilizando una función de prueba de la forma de\(\phi\), como se indicó anteriormente. La energía exacta calculada numéricamente del estado fundamental es\(0.808616 \hbar^{\frac{2}{3}}m^{-\frac{1}{3}}a^{-\frac{2}{3}}\). ¿Cuál es el porcentaje de error en tu valor?
Otro problema del método variacional
17. El oscilador armónico es especificado por el hamiltoniano:
\[H = -\dfrac{\hbar^2}{2m}\dfrac{d^2}{dx^2}+ \dfrac{1}{2}kx^2 ,\]
Supongamos que se desconoce la solución del estado base a este problema, y que se desea aproximarlo usando el teorema variacional. Elija como su función de onda de prueba,
\ [\ begin {array} {ll}
\ phi =\ sqrt {\ dfrac {15} {16}} a^ {-\ frac {5} {2}} &\ text {for} -a < x < a\\
\ phi = 0 &\ text {para} |x|\ ge a
\ end {array}\]
donde a es un parámetro arbitrario que especifica el rango de la función de onda. Tenga en cuenta que f se normaliza correctamente como se da.
a. Calcular\(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) y mostrarlo para ser dado por:
\[\int_{-\infty}^{\infty}\phi^*H\phi\, dx=\dfrac{5}{4}\dfrac{\hbar^2}{ma} + \dfrac{ka^2}{14}.\]
b. Calcular\(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) para\(a = b\Big(\dfrac{\hbar^2}{km}\Big)^{\frac{1}{4}}\).
c. Para encontrar la mejor aproximación a la verdadera función de onda y su energía, encontrar el mínimo de\(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) fijando\(\displaystyle\dfrac{d}{da}\int_{-\infty}^{\infty}\phi^*H\phi\, dx= 0\) y resolviendo para\(a\). Sustituir este valor en la expresión para\(\displaystyle\int_{-\infty}^{\infty}\phi^*H\phi\, dx\) dado en la parte a. para obtener la mejor aproximación para la energía del estado fundamental del oscilador armónico.
d. ¿Cuál es el porcentaje de error en su energía calculada de la parte c.?
Problema de la teoría de perturbación
18. Considera un electrón obligado a moverse sobre la superficie de una esfera de radio\(r_0\). El hamiltoniano para tal movimiento consiste únicamente en un término de energía cinética\(H_0 = \dfrac{L^2}{2m_er_0^2}\), donde\(L\) es el operador de momento angular orbital que involucra derivadas con respecto a las coordenadas polares esféricas (\(\theta,\phi\)). \(H_0\)tiene el conjunto completo de funciones propias\(\psi= Y_{l,m}(\theta,\phi)\).
a. Calcular los niveles de energía de orden cero de este sistema.
b. Se aplica un campo eléctrico uniforme a lo largo del\(z\) eje, introduciendo una perturbación\(V = -e\varepsilon z = -e\varepsilon r_0\cos\theta\), donde\(\varepsilon\) está la intensidad del campo. Evaluar la corrección a la energía del nivel más bajo a través de segundo orden en teoría de perturbación, utilizando la identidad
\[ \cos\theta Y_{l,m}(\theta,\phi) = \sqrt{\dfrac{(l+m+1)(l-m+1)}{(2l+1)(2l+3)}}Y_{l+1,m}(\theta,\phi) + \sqrt{\dfrac{(l+m)(l-m)}{(2l+1)(2l-1)}}Y_{l-1,m}(\theta,\phi) .\]
Tenga en cuenta que esta identidad le permite utilizar la ortonormalidad de los armónicos esféricos.
c. La polarizabilidad eléctrica\(\alpha\) da la respuesta de una molécula a un campo eléctrico aplicado externamente, y se define por\(\alpha = -\dfrac{\partial^2 E}{\partial \varepsilon^2}\) dónde\(E\) está la energía en presencia del campo y\(\varepsilon\) es la intensidad del campo. Calcular\(\alpha\) para este sistema.
d. Utilice este problema como modelo para estimar la polarizabilidad de un átomo de hidrógeno, donde\(r_0 = a_0 = 0.529\) Å, y un átomo de cesio, que tiene un solo electrón 6s con\(r_0 \approx 2.60 Å\). Los valores experimentales correspondientes son\(\alpha_H = 0.6668 Å^3\) y\(\alpha_{Cs} = 59.6 Å^3\).
Problema de Hartree-Fock que puedes hacer a mano
19. Dadas las siguientes energías orbitales (en hartrees) para el\(N\) átomo y los elementos de acoplamiento entre dos átomos similares (estos elementos de acoplamiento son los elementos de la matriz Fock a partir de cálculos estándar de SCF ab-initio minimum-basis), se calculan los niveles de energía orbitales moleculares y orbitales. Dibujar el diagrama de correlación orbital para la formación de la\(N_2\) molécula. Indicar la simetría de cada orbital atómico y molecular. Designe cada uno de los orbitales moleculares como enlazantes, no enlazantes o antiligantes.
\[N_{1s} = -15.31^*\]
\[ N_{2s} = -0.86^*\]
\[N_{2p} = -0.48^*\]
\(N_2\)\(\sigma_g\)Matriz de fock*
\ [\ left [\ begin {array} {ccc}
-6.52 & &\\
-6.22 & -7.06 &\\
3.61 & 4.00 & -3.92
\ end {array}\ derecha]\]
\(N_2\)\(\pi_g\)Matriz de fock*
\[[0.28]\]
\(N_2\)\(\sigma_u\)Matriz de fock*
\ [\ left [\ begin {array} {ccc}
1.02 & &\\
-0.60 & -7.59 &\\
0.02 & 7.42 & -8.53
\ end {array}\ derecha]\]
\(N_2\)\(\pi_u\)Matriz de fock*
\[-0.58\]
*Las matrices de Fock (y energías orbitales) se generaron usando cálculos de SCF estándar de conjunto de bases mínimas. Las matrices de Fock se encuentran en la base ortogonal formada a partir de estos orbitales.
problema de diagrama de correlación orbital
20. Dadas las siguientes energías orbitales de valencia para el\(C\) átomo y la\(H_2\) molécula, dibujar el diagrama de correlación orbital para la formación de la\(CH_2\) molécula (a través de una\(C_{2v}\)\(C\) inserción de en\(H_2\) resultando en flexión\(CH_2\)). Designar la simetría de cada orbital atómico y molecular tanto en su simetría de grupo puntual más alto como en la de la ruta de reacción (\(C_{2v}\)).
\ [\ begin {array} {ll}
C_ {1s} = -10.91^* & H_2\;\ sigma_g = -0.58^*\\
C_ {2s} = -0.60^* & H_2\;\ sigma_u = 0.67^*\\
C_ {2p} = -0.33^* &\ end {array}\]
*Las energías orbitales se generaron utilizando cálculos estándar de SCF de conjunto mínimo de bases STO3G.
Practicar usando simetría de grupo
21. Analizar cualitativamente la estructura electrónica (energías orbitales y orbitales) de\(PF_5\). Analizar solo los\(3p\) electrones\(3s\) y de P y el electrón de\(2p\) enlace de cada uno\(F\). Proceder con un\(D_{3h}\) análisis de la siguiente manera:
- Simetría adaptar los orbitales\(F\) atómicos superior e inferior.
- Simetría adaptar los tres orbitales\(F\) atómicos (trigonales).
- Simetría adaptar los orbitales P\(3s\) y\(3p\) atómicos.
- Permita que estos tres conjuntos de\(D_{3h}\) orbitales interactúen y dibuje el diagrama de energía orbital resultante.
- La simetría marca cada uno de estos niveles de energía molecular. Llena este diagrama de energía con 10 electrones de “valencia”.
Practica con símbolos de término y funciones de onda determinentales para átomos y moléculas
22. Para las ocupaciones orbitales dadas (configuraciones) de los siguientes sistemas, determinar todos los estados posibles (todas las combinaciones posibles permitidas de estados de espín y espacio). No hay necesidad de formar las funciones de onda determinentales, simplemente etiquetar cada estado con su propio símbolo de término.
\ [\ begin {array} {lll}
{\ rm a.} & CH_2 & 1a_1 {} ^2\ ,2a_1 {} ^2\ ,1b_2 {} ^2\ ,3a_1 {} ^1\ ,1b_1 {} ^1\ ,1b_1 {} ^1\\
{\ rm b.} & B_2 & 1\ sigma_g {} ^2\ ,1\ sigma_u {} ^2\ ,2\ sigma_g {} ^2\ ,2\ sigma_u {} ^2\ ,1\ pi_u {} ^1\ ,2\ pi_u {} ^1\\
{\ rm c.} & O_2 & 1\ sigma_g {} ^2\ ,1\ sigma_u {} ^2\ ,2\ sigma_g {} ^2\ ,2\ sigma_u {} ^2\ ,1\ pi_u {} ^4\ ,3\ sigma_g {} ^2\ ,1\ pi_g {} ^2\\
{\ rm d.} & Ti y 1s {} ^2\ ,2s {} ^2\ ,2p {} ^6\ ,3s {} ^2\ ,3p {} ^6\ ,4s {} ^2\ ,3d {} ^1\ ,4d {} ^1\\
{\ rm e.} & Ti y 1s {} ^2\ ,2s {} ^2\ ,2p {} ^6\ ,3s {} ^2\ ,3p {} ^6\ ,4s {} ^2\ ,3d {} ^2
\ end {array}\]
23. Construya funciones de onda determinantes de Slater para cada uno de los siguientes estados de\(CH_2\):
- \(^1B_1\)(\(1a_1{}^2\,2a_1{}^2\,1b_2{}^2\,3a_1{}^1\,1b_1{}^1\))
- \(^3B_1\)(\(1a_1{}^2\,2a_1{}^2\,1b_2{}^2\,3a_1{}^1\,1b_1{}^1\))
- \(^1A_1\)(\(1a_1{}^2\,2a_1{}^2\,1b_2{}^2\,3a_1{}^2\))
Woodward-Hoffmann gobierna el problema
24. Investiguemos las reacciones:
i.\(CH_2(^1A_1) \rightarrow H_2 + C\), y
ii. \(CH_2(^3B_1) \rightarrow H_2 + C\),
bajo una supuesta vía de\(C_{2v}\) reacción utilizando la siguiente información:
\(C\)átomo:\(^3P\)\(^1D\)\(^1S\)
\(C(^3P) + H_2 \rightarrow CH_2(^3B_1)\)\(\Delta E = -78.8\)kcal/mole
\(C(^1D) + H_2 \rightarrow CH_2(^1A_1)\)\(\Delta E = -97.0\)kcal/mole
IP (\(H_2\)) > IP (2s de carbono).
- Anote (primero en términos de\(2p_{1,0,-1}\) orbitales y luego en términos de\(2p_{x,y,z}\) orbitales) el:
- tres funciones de onda determinantes de Slater (SD) pertenecientes al\(^3P\) estado, todas las cuales tienen\(M_S = 1\),
- cinco funciones de onda\(^1D\) SD, y
- una función de onda\(^1S\) SD.
- Usando el sistema de coordenadas que se muestra a continuación, etiquetar los orbitales de hidrógeno sg, su y los orbitales de carbono\(2s\)\(2p_x\)\(2p_y\)\(2p_z\),,,, como\(a_1, b_1(x), b_(y),\) o\(a_2\). Haga lo mismo para los\(\sigma\),\(\sigma\)\(\sigma^*\),\(\sigma^*\),\(n\), y\(\pi_p\) orbitales de\(CH_2\).
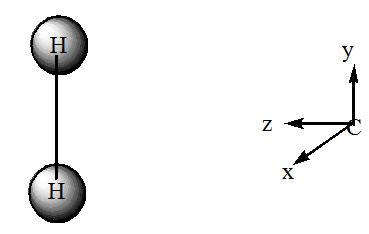
- Dibujar un diagrama de correlación orbital para las\(CH_2 \rightarrow H_2 + C\) reacciones. Tratar de representar correctamente los ordenamientos energéticos relativos de los orbitales.
- Dibuje un diagrama de correlación de configuración para\(CH_2(^3B_1) \rightarrow H_2 + C\) mostrar todas las configuraciones que surgen de los\(C(^3P) + H_2\) productos. Se puede suponer que las configuraciones doblemente excitadas se encuentran mucho (~100 kcal/mol) por encima de sus configuraciones padre.
- Repita el paso d. para\(CH_2(^1A_1) \rightarrow H_2 + C\) volver a mostrar todas las configuraciones que surgen de los\(C(^1D) + H_2 \) productos.
- ¿Esperas que la reacción\(C(^3P) + H_2 \rightarrow CH_2\) tenga una gran barrera de activación? ¿Acerca de qué tan grande? ¿Qué estado de\(CH_2\) se produce en esta reacción? ¿Se espera que las distorsiones alejadas de la\(C_{2v}\) simetría aumenten o bajen la barrera de activación? Mostrar cómo se podría estimar en qué parte de la ruta de reacción se produce la parte superior de la barrera.
- ¿\(C(^1D) + H_2 \rightarrow CH_2\)Se esperaría que tuviera una barrera mayor o menor de la que encontró para la\(^3P\)\(C\) reacción?
Otro problema de la regla Woodward-Hoffmann
25. La descomposición del carbeno singlete del estado tierra-estado,

para producir acetileno y\(^1D\) carbono se sabe que ocurre con una energía de activación igual a la endotermicidad de la reacción. Sin embargo, cuando el carbeno triplete correspondiente se descompone en acetileno y carbono en estado fundamental (triplete), la energía de activación excede la endotermicidad de esta reacción. Construya diagramas orbitales, de configuración y de correlación de estados que le permitan explicar las observaciones anteriores. Indicar si se requerirían funciones de onda de interacción de configuración única o configuración para describir los procesos de descomposición de singlete y triplete anteriores.
Práctica con espectrocopia rotacional y su relación con la estructura molecular
26. Considere las moléculas\(CCl_4\),\(CHCl_3\), y\(CH_2Cl_2\).
- Qué tipo de rotor son (parte superior simétrica, etc; no se moleste con oblato, o casi prolate, etc.)
- ¿Mostrarán espectros rotacionales puros (es decir, microondas)?
27. Supongamos que el amoníaco muestra un espectro rotacional puro. Si las constantes rotacionales son 9.44 y 6.20 cm -1, use la expresión energética:
\[E = (A - B) K^2 + B J(J + 1),\]
para calcular las energías (en cm -1) de las tres primeras líneas (es decir, aquellas con menor\(K\) número\(J\) cuántico para el nivel de absorción) en el espectro de absorción (ignorando términos de orden superior en la expresión energética).
problema en espectroscopía de vibración-rotación
28. La molécula\(^{11}B ^{16}O\) tiene una frecuencia vibracional\(\omega_e = 1885\) cm -1, una constante rotacional\(B_e = 1.78\) cm -1, y una energía de enlace desde el fondo del pozo potencial de\(D= 8.28\) eV. Utilice masas atómicas integrales en lo siguiente:
En la aproximación de que la molécula puede representarse como un oscilador Morse, calcular la longitud del enlace,\(R_e\) en angstroms, la constante de distorsión centrífuga,\(D_e\) en cm -1, la constante de anarmonicidad,\(\omega_e x_e\) en cm -1, la energía de enlace corregida en punto cero, \(D_0^0\)en eV, la constante de interacción de rotación de vibración,\(\alpha_e\) en cm -1, y las constantes de rotación específicas del estado vibracional,\(B_0\) y\(B_1\) en cm -1. Utilice la expresión de energía de vibración-rotación para un oscilador Morse:
\[E = \hbar\omega_e\Big(v + \dfrac{1}{2}\Big) - \hbar\omega_e x_e\Big(v + \dfrac{1}{2}\Big)^2 + B_vJ(J + 1) - D_eJ^2(J + 1)^2,\]donde
\[Bv = B_e - \alpha_e\Big(v + \dfrac{1}{2}\Big),\; \alpha_e = \dfrac{-6B_e^2}{\hbar\omega_e}+ \dfrac{6\sqrt{B_e^3\hbar\omega_e x_e}}{\hbar\omega}, \; \text{and } D_e = \dfrac{4B_e^3}{\hbar\omega^2}.\]
¿Esta molécula mostrará un espectro de rotación puro? ¿Un espectro de vibración-rotación? Suponiendo que sí, ¿cuáles son las energías (en cm -1) de las tres primeras líneas en la rama P (\(\Delta v = +1, \Delta J = -1\)) de la absorción fundamental?
problema etiquetando modos vibracionales por simetría
29. Considera trans -\(C_2H_2Cl_2\). Se muestran los modos vibracionales normales de esta molécula
abajo. ¿Cuál es la simetría de la molécula? Etiquetar cada uno de los modos con la representación irreducible apropiada.
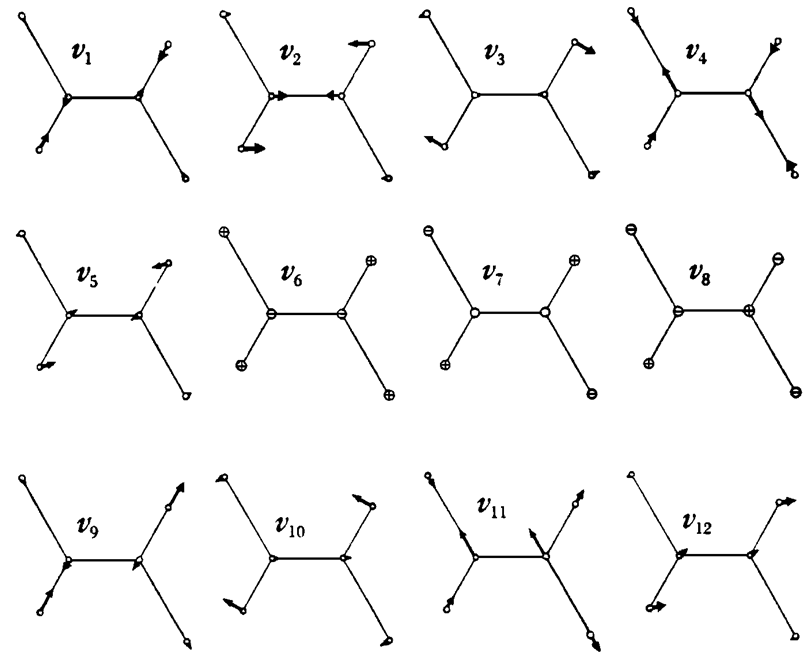
problema en espectroscopía rotacional
30. Supongamos que se le dan dos moléculas (una es\(CH_2\) y la otra es\(CH_2^-\) pero no sabe cuál es cuál). Ambas moléculas tienen\(C_{2v}\) simetría. La longitud del\(CH\) enlace de la molécula I es 1.121 Å y para la molécula II es 1.076 Å. El ángulo de enlace de la molécula I es 104° y para la molécula II es 136°.
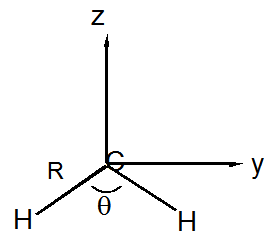
a. Utilizando un sistema de coordenadas centrado en el\(C\) núcleo como se muestra anteriormente (la molécula está en el\(YZ\) plano), computar el momento de inercia tensores de ambas especies (I y II). Las definiciones de los componentes del tensor son, por ejemplo:
\[I_{xx} = - \sum_j m_j (y_j^2+z_j^2) - M(Y^2 + Z^2)\]
\[I_{xy} = -\sum_j m_j x_j y_j - MXY\]
Aquí,\(m_j\) está la masa del núcleo\(j\),\(M\) es la masa de toda la molécula, y\(X\),\(Y\),\(Z\) son las coordenadas del centro de masa de la molécula. Use Å para distancias y amu para masas.
b. Encontrar los principales momentos de inercia\(I_a < I_b < I_c\) para ambos compuestos (en unidades amu Å 2) y convertir estos valores en constantes rotacionales\(A\)\(B\), y\(C\) en cm -1 usando, por ejemplo,
\[A = \dfrac{h}{8\pi^2cI_a}.\]
c. Ambos compuestos son “picos casi prolatados” cuyos niveles de energía pueden aproximarse bien usando la fórmula superior de prolato:
\[E = (A - B) K^2 + BJ(J + 1),\]
si se utiliza para la\(B\) constante el promedio de la\(B\) y\(C\) valorada determinada anteriormente. Así, tomar los\(C\) valores\(B\) y (para cada compuesto) y promediarlos para producir una\(B\) constante efectiva para usar en la fórmula energética anterior. Anote (en unidades cm -1) la fórmula energética para ambas especies. ¿Qué valores son\(J\) y\(K\) se les permite asumir? ¿Cuál es la degeneración del nivel etiquetado por un dado\(J\) y\(K\)?
d. Dibuje una imagen de ambos compuestos y muestre las direcciones de los tres ejes principales (a, b, c). En estas imágenes, se muestra el tipo de movimiento de rotación asociado con el número cuántico\(K\).
e. Supongamos que se le da el espectro fotoelectrónico de\(CH_2^-\). En este espectro\(J_j = J_i + 1\) las transiciones se denominan absorciones de ramificación R y las que obedecen\(J_j = J_i - 1\) se denominan transiciones de ramificación P. El espaciado entre líneas puede aumentar o disminuir como funciones de\(J_i\) dependiendo de los cambios en el momento de inercia para la transición. Si los espaciamientos se acercan cada vez más, decimos que el espectro exhibe una llamada formación de cabeza de banda. En el espectro fotoelectrónico que se le da, se realiza un análisis rotacional de las líneas vibracionales en este espectro y se encuentra que las ramas R muestran formación de cabeza de banda pero las ramas P no. Con base en esta información, determinar qué compuesto I o II es el\(CH_2^-\) anión. Explícale razonamiento.
f. ¿A qué\(J\) valor (de la especie absorbente) se produce la cabeza de banda y a qué diferencia de energía rotacional?
Uso de simetría de grupo puntual en espectroscopía vibracional
31. Consideremos los movimientos vibracionales del benceno. Para considerar todos los modos vibracionales del benceno debemos adjuntar un conjunto de vectores de desplazamiento en el\(x\),\(y\), y\(z\) direcciones a cada átomo en la molécula (dando 36 vectores en total), y evaluar cómo estos se transforman bajo las operaciones de simetría de\(D_{6h}\). Para este problema, sin embargo, solo indagemos sobre las vibraciones de\(C-H\) estiramiento.
a. Representar el movimiento de\(C-H\) estiramiento en cada\(C-H\) enlace mediante un vector dirigido hacia afuera en cada\(H\) átomo, designado\(r_i\):
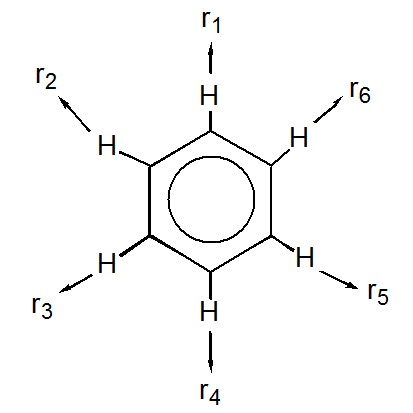
b. Estos vectores forman la base de una representación reducible. Evaluar los caracteres para esta representación reducible bajo las operaciones de simetría del\(D_{6h}\) grupo.
c. Descomponer la representación reducible obtenida en la parte b. en sus componentes irreducibles. Estas son las simetrías de los diversos modos vibratorios de\(C-H\) estiramiento en benceno.
d. El estado vibracional con cuantos cero en cada uno de los modos vibracionales (el estado vibracional del suelo) de cualquier molécula siempre pertenece a la representación totalmente simétrica. Para el benceno, el estado vibratorio del suelo es, por tanto, de\(A_{1g}\) simetría. Un estado excitado que tiene un cuántico de excitación vibracional en un modo que es de una especie de simetría dada tiene la misma especie de simetría que el modo que está excitado (porque las funciones de onda vibracional se dan como polinomios hermitas en la coordenada de estiramiento). Así, por ejemplo, la excitación (por un cuántico) de un modo vibracional de\(A_{2u}\) simetría da una función de onda de\(A_{2u}\) simetría. Para resolver la cuestión de qué modos vibracionales pueden ser excitados por la absorción de la radiación infrarroja debemos examinar los\(x\),\(y\), y\(z\) componentes de la integral del dipolo de transición para las funciones de onda de estado inicial y final\(\psi_i\) y\(\psi_f\), respectivamente:
\[|\langle\psi_f | x |\psi_i \rangle| ,\;\; |\langle\psi_f | y |\psi_i \rangle| ,\; \; \text{and } |\langle\psi_f | z |\psi_i \rangle| .\]
Utilizando la información proporcionada anteriormente, ¿cuál de los modos\(C-H\) vibracionales del benceno será infraro-activo y cómo se polarizarán las transiciones? ¿Cuántas\(C-H\) vibraciones observarás en el espectro infrarrojo del benceno?
e. Un modo vibracional estará activo en la espectroscopia Raman solo si una o más de las siguientes integrales no son cero:
\[|\langle\psi_f | xy |\psi_i \rangle| ,\;\;|\langle\psi_f | xz |\psi_i \rangle| , \;\; |\langle\psi_f | yz |\psi_i \rangle| ,\]
\[|\langle\psi_f | x^2 |\psi_i \rangle| ,\;\; |\langle\psi_f | y^2 |\psi_i \rangle| ,\;\; \text{and } |\langle\psi_f | z^2 |\psi_i \rangle| .\]
Utilizando el hecho de que los operadores cuadráticos se transforman de acuerdo con las representaciones irreducibles:
\[(x^2 + y^2, z^2) \Rightarrow A_{1g}\]
\[(xz, yz) \Rightarrow E_{1g}\]
\[(x^2 - y^2, xy) \Rightarrow E_{2g}\]
Determinar cuál de los modos\(C-H\) vibracionales será Raman activo.
f. ¿Hay alguno de los movimientos vibratorios de\(C-H\) estiramiento del benceno que no se pueda observar en ninguno de los infrarrojos de la espectroscopia Raman? Dar la etiqueta de representación irreducible para estos modos no observables.
problema en espectros electrónicos y tiempos de vida
32. La teoría de perturbación dependiente del tiempo proporciona una expresión para la vida radiativa de un estado electrónico excitado, dada por\(t_R\):
\[t_R = \dfrac{3\hbar^4c^3}{4(E_i-E_f)^3|\mu_{fi}|^2},\]
donde\(i\) se refiere al estado excitado,\(f\) se refiere al estado inferior, y\(\mu_{fi}\) es el dipolo de transición.
a. Evaluar el\(z\) componente -del dipolo de transición para la\(2p_z \rightarrow 1s\) transición en un átomo hidrogénico de carga nuclear\(Z\), dado:
\[\psi_{1s} =\dfrac{1}{\sqrt{\pi}} \bigg(\dfrac{Z}{a_0}\bigg)^{\frac{3}{2}} \exp\bigg(-\dfrac{Zr}{a_0}\bigg) ,\text{ and } \psi_{2p_z} =\dfrac{1}{4\sqrt{2\pi}} \bigg(\dfrac{Z}{a_0}\bigg)^{\frac{5}{2}} r \cos\theta\exp\bigg(-\dfrac{Zr}{2a_0}\bigg).\]
Exprese su respuesta en unidades de\(ea_0\).
b. Utilice la simetría para demostrar que los\(y\) componentes\(x-\) y de\(\mu_{fi}\) son cero, i.e.
\[\langle 2p_z| e x |1s\rangle = \langle 2p_z| e y |1s\rangle = 0.\]
c. Calcular la vida radiativa\(t_R\) de un átomo similar a hidrógeno en su\(2p_z\) estado. Usa la relación\(e^2 =\dfrac{\hbar^2}{m_ea_0}\) para simplificar tus resultados.
diferencia entre activar lenta y rápidamente una perturbación
33. Consideremos un caso en el que se conozca el conjunto completo de estados {\(\phi_k\)} para un hamiltoniano.
a. si el sistema se encuentra inicialmente en el estado m en el\(t=0\) momento en que\(V\) se enciende repentinamente una perturbación constante, encuentre las amplitudes de probabilidad\(C_k^{(2)}(t)\) y\(C_m^{(2)}(t)\), a segundo orden en\(V\), que describan que el sistema está en un estado diferente\(k\) o en el mismo estado \(m\)en el momento\(t\).
b. Si la perturbación se enciende adiabáticamente (es decir, muy lentamente), ¿qué son\(C_k^{(2)}(t)\) y\(C_m^{(2)}(t)\)? Aquí, considere que el tiempo inicial es\(t_0 \rightarrow -\infty\), y el potencial es\(V e^\eta t\), donde se permite que el parámetro positivo\(\eta\) se acerque a cero\(\eta\rightarrow 0\) para describir la perturbación adiabáticamente encendida.
c. Comparar los resultados de las partes a. y b. y explicar las diferencias.
d. Ignorar las contribuciones de primer orden (suponga que desaparecen) y evaluar las tasas de transición
\(|C_k^{(2)}(t)|^2\)para los resultados de la parte b. tomando el límite\(\eta \rightarrow 0^+\), para obtener los resultados adiabáticos.
ejemplo de encender rápidamente una perturbación- la aproximación repentina
34. Considere una interacción o perturbación que se lleva a cabo repentinamente (instantáneamente, por ejemplo, dentro de un intervalo de tiempo\(\Delta t\) que es pequeño en comparación con el período natural\(\omega_{nm}^{-1}\) correspondiente a la transición de estado\(m\) a estado\(n\)), y después de eso se apaga adiabáticamente ( es decir, extremadamente lentamente como\(V e^\eta t\)). La probabilidad de transición en este caso se da como:
\[T_{nm} \approx \dfrac{|\langle n|V|m \rangle|^2}{\hbar^2\omega_{nm}^2}\]
donde\(V\) corresponde al valor máximo de la interacción cuando se enciende. Esta fórmula permite calcular las probabilidades de transición bajo la acción de perturbaciones repentinas que son pequeñas en valor absoluto siempre que la teoría de perturbaciones sea aplicable.
Usemos esta “aproximación repentina” para calcular la probabilidad de excitación de un electrón bajo un cambio repentino de la carga del núcleo. Considera la reacción:
\[^3_1H \rightarrow ^3_2He^+ + e^-,\]
y supongamos que el átomo de tritio tiene su electrón inicialmente en un\(1s\) orbital.
a. Calcular la probabilidad de transición para la transición\(1s \rightarrow 2s\) para esta reacción usando la fórmula anterior para la probabilidad de transición.
b. Supongamos que en\(t = 0\) el momento el sistema se encuentra en un estado que corresponde al
función de onda\(\phi_m\), que es una función propia del operador\(H_0\). A\(t = 0\), se produce el cambio repentino del hamiltoniano (ahora denotado como\(H\) y permanece sin cambios). Calcular la misma probabilidad de\(1s \rightarrow 2s\) transición que en la parte a., solo que esta vez como el cuadrado de la magnitud del coeficiente,\(A_{1s,2s}\) utilizando la expansión:
\[\Psi(r,0) =\phi_m(r) =\sum_n A_{nm}\psi_n(r) ,\]donde\[A_{nm} =\int \phi_m(r)\psi_n(r)d^3r\]
Tenga en cuenta que las funciones propias de\(H\) son\(\psi_n\) con valores propios\(E_n\). Compare este valor con el obtenido por la teoría de perturbaciones en la parte a.
problema simétrico del espectro rotacional superior
35. La molécula de yoduro de metilo se estudia mediante microondas (rotacional puro)
espectroscopía. La siguiente integral gobierna las reglas de selección rotacional para las transiciones etiquetadas\(J, M, K \rightarrow J', M', K'\):
\[I = <D_{M'K'}^{J'}|\boldsymbol{\varepsilon}\bullet\boldsymbol{\mu}|D_{MK}^J>.\]
El momento dipolar\(\boldsymbol{\mu}\) se encuentra a lo largo del eje de\(C_3\) simetría de la molécula. Deje que el campo eléctrico de la luz\(\boldsymbol{\mu}\) defina la dirección Z fija en el laboratorio.
a. Utilizando el hecho de que\(\cos\beta = D_{00}^*\), demostrar que
\[I = 8\pi^2\mu\varepsilon(-1)^{(M+K)} \left(\begin{array}{ccc}J' & 1 & J \\ M & 0 & M \end{array}\right) \left(\begin{array}{ccc}J' & 1 & J \\ K & 0 & K \end{array}\right) \delta_{M'M}\delta_{K'K}\]
b. ¿Qué restricciones impone este resultado\(\Delta J = J' - J\)? Explique físicamente por qué el número\(K\) cuántico no puede cambiar.
problema en espectroscopía electrónica y fotoelectrónica
36. Considera la molécula\(BO\).
a. ¿Cuál es el número total de estados electrónicos posibles que se pueden formar por combinación de estado fundamental\(B\) y\(O\) átomos?
b. ¿Qué configuraciones electrónicas de la molécula probablemente sean bajas en energía? Considerar todos los ordenamientos razonables de los orbitales moleculares. ¿Cuáles son los estados correspondientes a estas configuraciones?
c. ¿Cuáles son las órdenes de fianza en cada uno de estos estados?
d. El verdadero estado fundamental de\(BO\) es\(^2\Sigma\). Especifique las simetrías +/- y u/g para este estado.
e. ¿Cuál de los estados excitados que derivó anteriormente irradiará al estado\(^2\Sigma\) base? Considera solo la radiación dipolo eléctrico.
f. ¿La ionización de la molécula para formar un catión conduce a una fuerza de enlace más fuerte, más débil o equivalente?
g. Suponiendo que las energías de los orbitales moleculares no cambian con la ionización, ¿cuáles son el estado fundamental, el primer estado excitado y el segundo estado excitado del ion positivo?
h. Considerando únicamente estos estados, predecir la estructura del espectro fotoelectrónico que obtendría para la ionización de\(BO\).
problema en espectroscopía de vibración-rotación
37.
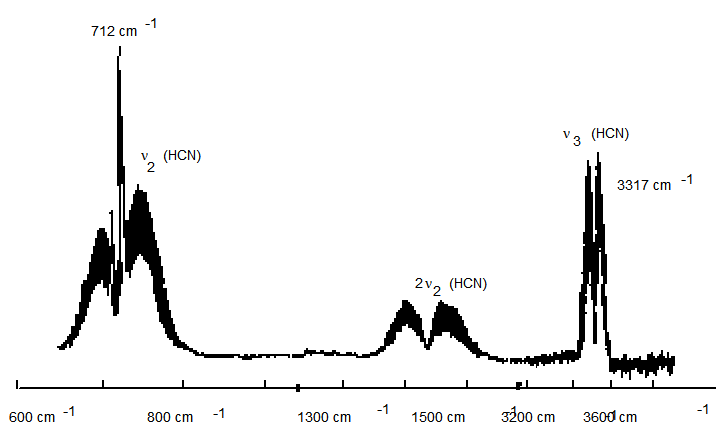
La figura anterior muestra parte del espectro de absorción infrarroja del\(HCN\) gas. La molécula tiene una vibración de\(CH\) estiramiento, una vibración de flexión y una vibración de\(CN\) estiramiento.
a. ¿Alguna de las vibraciones de lineal es\(HCN\) degenerada?
b. ¿A qué vibración pertenece el grupo de picos entre 600 cm -1 y 800 cm -1?
c. ¿A qué vibración pertenece el grupo de picos entre 3200 cm -1 y 3400 cm -1?
d. ¿Cuáles son las simetrías (s, p, d) del\(CH\) estiramiento,\(CN\) estiramiento y flexión
movimientos vibracionales?
e. Comenzando con\(HCN\) en su nivel vibracional 0,0,0, cuyas transiciones fundamentales serían activas por infrarrojos bajo luz polarizada paralela (es decir, polarización del eje z):
\[000 \rightarrow 001?\]
\[000 \rightarrow 100?\]
\[000 \rightarrow010?\]
f. ¿Por qué la transición 712 cm -1 tiene una rama Q, mientras que cerca de 3317 cm -1 solo tiene ramas P- y R?
Problema en el que puedes practicar derivar ecuaciones
Esto es importante porque un científico teórico hace derivaciones como parte de su trabajo
38.
Al expandir los orbitales moleculares {\(\phi_k\)} como combinaciones lineales de orbitales atómicos {\(\chi_\mu\)},
\[\phi_k =\sum_\mu c_{\mu k}\chi_\mu\]
muestran cómo las ecuaciones canónicas de Hartree-Fock (HF):
\[F \phi_i = \varepsilon_i \phi_j\]
reducir a la ecuación de tipo de valor propio de la matriz de la forma:
\[\sum_\nu F_{\mu\nu}C_{\nu i}= \varepsilon_i\sum_\nu S_{\mu\nu}C_{\nu i}\]
donde:
\[F_{\mu\nu} = \langle \chi_\mu|h|\chi_nu\rangle+\sum_{\delta\kappa} \left[\gamma_{\delta\kappa}\langle\chi_\mu\chi_\delta|g|\chi_\nu\chi_\kappa\rangle - \gamma_{\delta\kappa}^{\rm ex} \langle\chi_\mu\chi_\delta|g|\chi_\kappa\chi_\nu\rangle\right] ,\]
\[S_{\mu\nu} = \langle\chi_\mu|\chi_nu\rangle,\; \gamma_{\delta\kappa} =\sum_{i={\rm occ}}C_{\delta i}C_{\kappa i} ,\]
y
\[\gamma_{\delta\kappa}^{\rm ex} =\sum_{i=\text{occ and same spin}} C_{\delta i}C_{\kappa i}.\]
Tenga en cuenta que la suma sobre\(i\) en\(\gamma_{\delta\kappa}\) y\(\gamma_{\delta\kappa}^{\rm ex}\) es una suma sobre los orbitales de giro. Además, demuestre que esta matriz de Fock se puede reducir aún más para la carcasa cerrada para:
\[F_{\mu\nu} = \langle \chi_\mu|h|\chi_nu\rangle +\sum_{\delta\kappa} P_{\delta\kappa}\bigg[\langle\chi_\mu\chi_\delta|g|\chi_\nu\chi_\kappa\rangle - \dfrac{1}{2} \langle\chi_\mu\chi_\delta|g|\chi_\kappa\chi_\nu\rangle\bigg] ,\]
donde la matriz de orden de bonos de carga\(P\),, se define como:
\[P_{\delta\kappa} =\sum_{i={\rm occ}} 2C_{\delta i}C_{\kappa i},\]
donde la suma de\(i\) aquí es una suma sobre orbitales no orbitales giratorios.
Otro problema de práctica de derivación
39. Mostrar que la energía total de HF para un sistema de carcasa cerrada puede escribirse en términos de integrales sobre los orbitales de HF ortonormales como:
\[E({\rm SCF}) = 2\sum_k^{\rm occ}\langle\phi_k|h|\phi_k\rangle+\sum_{k,l}^{\rm occ} [2\langle kl|g|kl \rangle - \langle kl|g|lk\rangle] +\sum_{\mu>\nu} \dfrac{Z_\mu Z_\nu}{Z_{\mu\nu}}\]
.
Más problema de derivación
40. Demostrar que la energía total de HF puede expresarse alternativamente como:
\[E({\rm SCF}) =\sum_k^{\rm occ}(\varepsilon_k+\langle\phi_k|h|\phi_k\rangle)+ \sum_{\mu>\nu}\dfrac{Z_\mu Z_\nu}{R_{\mu\nu}},\]
donde se\(\varepsilon_k\) refieren a las energías orbitales de HF.
Problema Molecular Hartree-Fock SCF
41. Este problema estará relacionado con la realización de un cálculo SCF para la\(HeH^+\) molécula en estado\(1\Sigma_g^+(^1\sigma^2)\) fundamental. Las integrales de uno y dos electrones (en unidades atómicas) necesarias para realizar este cálculo de SCF a\(R = 1.4\) a.u. usando orbitales tipo Slater con exponentes orbitales de 1.6875 y 1.0 para el\(He\) y\(H\), respectivamente, son:
| S 11 = 1.0, | S 22 = 1.0, | S 12 = 0.5784 |
| h 11 = -2.6442, | h 22 = -1.7201, | h 12 = -1.5113, |
| g 1111 = 1.0547, | g 1121 = 0.4744, | g 1212 = 0.5664, |
| g 2211 = 0.2469, | g 2221 = 0.3504, | g 2222 = 0.6250, |
donde 1 se refiere\(1s_{He}\) y 2 a\(1s_H\). Las integrales de dos electrones se dan en notación Dirac. Las partes a. — d deben hacerse a mano. Cualquier pieza posterior puede hacer uso del software QMIC que se puede encontrar en
www.emsl.pnl.gov:2080/people/... a_nichols.html.
- Usando\(\phi_1 \approx 1s_{He}\) para la estimación inicial del orbital molecular ocupado, formar una matriz de 2x2 Fock. Utilice la ecuación derivada anteriormente en el problema 38 para\(F_{\mu\nu}\).
- Resolver las ecuaciones de valores propios de la matriz de Fock dadas anteriormente para obtener las energías orbitales y un orbital molecular ocupado mejorado. Al hacerlo, tenga en cuenta que\(\langle\phi_1|\phi_1\rangle= 1 = C_1^TSC_1\) da la condición de normalización necesaria para los coeficientes de expansión de\(\phi_1\) la base orbital atómica.
- Determinar la energía SCF total usando la expresión del problema 39 en esta etapa del procedimiento iterativo. ¿Cuándo coincidirá esta energía con la obtenida usando la expresión alternativa para\(E({\rm SCF})\) dada en el problema 40?
- Obtener el nuevo orbital molecular,\(\phi_1\), a partir de la solución del problema del valor propio de la matriz (parte b).
- Se puede obtener una nueva matriz de Fock y energía total relacionada con esta elección mejorada de orbital molecular,\(\phi_1\). Este proceso puede continuar hasta que se haya satisfecho un criterio de convergencia. Los criterios de convergencia típicos incluyen: ningún cambio significativo en los orbitales moleculares o la energía total (o ambas) de una iteración a la siguiente. Realice este procedimiento iterativo para el\(HeH^+\) sistema hasta que la diferencia de energía total entre dos iteraciones sucesivas sea inferior a 10 -5 a.u.
- Demostrar, comparando la diferencia entre la energía total SCF en una iteración y la energía total SCF convergente, que la convergencia del enfoque SCF anterior es principalmente lineal (o de primer orden).
- ¿La energía total SCF se calcula en cada iteración del procedimiento SCF anterior, ya que en el problema 39 es un límite superior a la energía total exacta del estado fundamental?
- ¿Esta función de onda SCF da lugar (at\(R\rightarrow\infty\)) a productos de disociación adecuados?
Problema de interacción de configuración
42. Este problema continuará abordando el mismo sistema\(HeH^+\) molecular que el anterior, extendiendo el análisis para incluir efectos de correlación. Utilizaremos las integrales de uno y dos electrones (misma geometría) en la base orbital molecular SCF convergente (a 10 -5 au) que habríamos obtenido después de 7 iteraciones anteriores. Los MO convergentes que debiste haber obtenido en el problema 1 son:
\[\phi_1 = \left[\begin{array}{c}-0.89997792\\-0.15843012\end{array}\right] \phi_2 =\left[\begin{array}{c}-0.83233180\\1.21558030\end{array}\right] \]
a. Realizar un cálculo de CI de dos configuraciones usando las\(2\sigma^2\) configuraciones\(1\sigma^2\) y primero obteniendo una expresión para los elementos de la matriz CI\(H_{I,J}\) (\(I,J = 1\sigma^2, 2\sigma^2\)) en términos de integrales de uno y dos electrones, y en segundo lugar mostrando que la matriz de CI resultante es (ignorando la nuclear energía de repulsión):
\[\left[\begin{array}{cc}-4.2720 & 0.1261 \\ 0.1261 & -2.0149\end{array}\right]\]
b. Obtener las dos energías de CI y vectores propios para la matriz que se encuentra en la parte a.
c. Demostrar que la función de onda CI de menor energía es equivalente a la siguiente función de onda de dos determinantes (configuración única):
\[\dfrac{1}{2}\left\{|(\sqrt{a}\phi_1 + \sqrt{b}\phi_2) \alpha(\sqrt{a}\phi_1 - \sqrt{b}\phi_2)\beta| + |(\sqrt{a}\phi_1 - \sqrt{b}\phi_2)\alpha (\sqrt{a}\phi_1 + \sqrt{b}\phi_2)\beta|\right\}\]
involucrando los orbitales polarizados:\(\sqrt{a}\phi_1 ± \sqrt{b}\phi_2\), dónde\(a = 0.9984\) y\(b = 0.0556\).
d. Amplíe la lista de CI a 3 configuraciones agregando\(1\sigma 2\sigma\) al original\(1\sigma^2\) y\(2\sigma^2\) las configuraciones de la parte a anterior. Primero, exprese la\(1\sigma 2\sigma\) configuración apropiada de espin-coupled singlete como una combinación de determinantes Slater y luego compute todos los elementos de esta matriz 3x3.
\(E\). Obtener todas las energías propias y los vectores propios normalizados correspondientes para este problema de CI.
f. Determinar las energías de excitación y los momentos de transición para\(HeH^+\) usar el resultado completo de CI de la parte e anterior. Los elementos de matriz que no se desvanecen del operador dipolo\(\textbf{r}(x,y,z)\) en la base atómica son:
\[\langle1s_H|z|1s_{He}\rangle= 0.2854 \text{ and } \langle1s_H|z|1s_H\rangle= 1.4.\]
Primero determinar los elementos de la matriz\(\textbf{r}\) en la base orbital SCF luego determinar las energías de excitación y los momentos de transición desde el estado fundamental a los dos estados singlete excitados de\(HeH^+\).
g. Pasando ahora a la teoría de la perturbación, llevar a cabo un cálculo de teoría de perturbación de la función de onda de primer orden\(|1\sigma^2\rangle^{(1)}\) para el caso en el que la función de onda de orden cero se toma como determinante de\(1\sigma^2\) Slater. Demuestre que la función de onda de primer orden viene dada por:
\[|1\sigma^2\rangle^{(1)} = -0.0442| 2\sigma^2\rangle.\]
h. ¿Por qué la\(|1\sigma2\sigma\rangle\) configuración no entra en la función de onda de primer orden?
i. Normalizar la función de onda resultante que contiene partes de orden cero más primer orden y compararla con la función de onda obtenida en el estudio CI de dos configuraciones de la parte b.
j. Mostrar que la energía de correlación RSPT de segundo orden\(E^{(2)}\), de\(HeH^+\) es -0.0056 a.u. ¿Cómo se compara esto con la energía de correlación obtenida del estudio de CI de dos configuraciones de la parte b?
Repitiendo el problema SCF pero con un programa de computadora
43. Usando cualquiera de los programas que están disponibles para usted o los programas QMIC que puede encontrar en el sitio web
www.emsl.pnl.gov:2080/people/... ichols_ja.html
calcular la energía SCF de\(HeH^+\) usar la misma geometría que en el problema 42 y el conjunto de bases STO3G proporcionado en la biblioteca de conjuntos de bases QMIC. ¿Cómo se compara esta energía con la encontrada en el problema 42? Vuelva a ejecutar el cálculo con la base 3-21G proporcionada. ¿Cómo se compara esta energía con el STO3G y la energía encontrada usando STOs en el problema 42?
Serie de cálculos SCF para producir una curva de energía potencial
44. Genere superficies de energía potencial SCF para\(HeH^+\) y\(H_2\) usando el software QMIC o sus propios programas. Utilice el conjunto de bases 3-21G y genere puntos para geometrías de\(R = 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.5,\) y\(10.0 a_0\). Trazar las energías vs. geometría para cada sistema. ¿Qué sistema se disocia correctamente?
Curvas de potencial de interacción de configuración para varios estados
45. Generar superficies de energía potencial CI para los 4 estados de\(H_2\) resultado de un cálculo con 2 electrones ocupando los 2 orbitales SCF más bajos (\(1\sigma_g\)y\(1\sigma_u\)) de todas las formas posibles. Utilice las mismas geometrías y bases que en el problema 44. Trazar las energías vs. geometría para cada sistema. Marcar y caracterizar adecuadamente cada uno de los estados (e.g., repulsivo, disociarse adecuadamente, etc.).
Problema en las funciones de partición y las propiedades termodinámicas
46. Los átomos F tienen estados electrónicos\(1s^2\,2s^2\,2p^5\)\(^2P\) terrestres que se dividen por acoplamiento espín-órbita\(^2P_{3/2}\) y\(^2P_{1/2}\) estados que difieren en solo 0.05 eV en energía.
a. escribir la función de partición electrónica (tomar la energía del\(^2P_{3/2}\) estado para ser cero y la del\(^2P_{1/2}\) estado para ser 0.05eV e ignorar todos los demás estados) para cada átomo F.
b. Utilizando, derivar una expresión para la energía electrónica promedio de átomos F\(N\) gaseosos.
c. Utilizando el hecho de que\(kT=0.03\) eV a\(T=300\) °K, hacer una gráfica (cualitativa) de\(\bar{E}/N\) vs\(T\) para\(T\) que van de 100°K a 3000°K.
Problema al usar la teoría del estado de transición
47. Supongamos que usamos la teoría del estado de transición para estudiar la reacción
\(NO(g) + Cl_2(g) \rightarrow NOCl(g) + Cl(g)\)suponiendo que procediera a través de un estado de transición doblada, y obtuvimos una expresión para el coeficiente de tasa
\[k_{\rm bent}=\dfrac{kT}{h}e^{-E^\ne/kT}\dfrac{\dfrac{q^\ne}{v}}{\dfrac{q_{NO}}{v} \dfrac{q_{Cl_2}}{v}}\]
a. ahora, consideremos qué diferencias se producirían si la estructura del estado de transición fuera lineal en lugar de doblada. Suponiendo que la energía de activación\(E^\ne\) y las degeneraciones del estado electrónico no se alteren, derivar una expresión para la relación de los coeficientes de tasa para los casos de estado de transición lineal y doblado
\[\dfrac{k_{\rm linear}}{k_{\rm bent}}\]
b. Usando las siguientes estimaciones de orden de magnitud de las funciones de partición traslacional, rotacional y vibracional por grado de libertad a 300°K
\[q_t \sim 10^8, q_r \sim 10^2, q_v \sim 1,\]
¿Cuál es la relación que esperarías\(k_{\rm linear}/k_{\rm bent}\)?
Problema con los determinantes de Slater
48. Mostrar que la configuración (determinante) correspondiente al\(Li^+\)\(1s(\alpha)1s(\alpha)\) estado se desvanece.
Otro problema con los determinantes de Slater y Momenta Angular
49. Construir las 3 funciones de onda triplete y 1 singlete para la\(Li^+\)\(1s^12s^1\) configuración. Mostrar que cada estado es una función propia apropiada de S2 y Sz (use operadores de elevación y descenso para S2)
Problema con los determinantes de Slater para una molécula lineal
50. Construir funciones de onda determinantes para cada estado de la\(1\sigma^22\sigma^23\sigma^21\pi^2\) configuración de\(NH\).
Problema con los determinantes de Slater para un átomo
51. Construir funciones de onda determinantes para cada estado de la\(1s^12s^13s^1\) configuración de\(Li\).
Problema en el Momento Angular de un Átomo
52. Determinar todos los símbolos de término que surgen de la\(1s^22s^22p^23d^1\) configuración del\(N\) átomo excitado.
Practica con las Reglas del Condón de Slater
53. Calcular la energía (usando las reglas del Condón de Slater) asociada a los electrones de\(2p\) valencia para los siguientes estados del\(C\) átomo.
i.\(^3P(M_L=1,M_S=1),\)
ii. \(^3P(M_L=0,M_S=0),\)
iii. \(^1S(M_L=0,M_S=0)\), y
iv. \(^1D(M_L=0,M_S=0)\).
Más práctica con las reglas del condón de Slater
54. Calcular la energía (usando las reglas del Condón de Slater) asociada a los electrones de\(\pi\) valencia para los siguientes estados de la\(NH\) molécula.
i.\(^1\Delta\)\((M_L=2, M_S=0),\)
ii. \(^1\Sigma\)\((M_L=0, M_S=0),\)y
iii. \(^3\Sigma\)\((M_L=0, M_S=0).\)
Practica con las ecuaciones de la mecánica estadística
55. Coincidir cada una de las ecuaciones siguientes con la frase adecuada A-K
\ [\ renovarcomando {\ arraystretch} {2.5}\ begin {array} {|c|c|}\ hline
B_2=-2\ pi\ int_0^\ infty r^2\ bigg (\ exp\ bigg (-\ dfrac {u (r)} {kT}\ bigg) -1\ bigg) dr &\ phantom {\ Bigg|}\ hspace {2cm}\\\ hline
\ bar {E^2} - (\ bar {E}) ^2=KT^2\ Grande (\ dfrac {\ parcial E} {\ parcial T}\ Grande) _ {N, V} &\ fantasma {\ Bigg|}\\\ hline
\ dfrac {2\ pi MKt} {h^2} &\ phantom {\ Bigg|}\\ hline
Q=\ exp\ bigg (-\ dfrac {N\ phi} {2kt}\ bigg)\ bigg (\ dfrac {\ exp (-\ theta/2t)} {1-\ exp (-\ theta/2t)} bigg) ^ {3N} &\ fantasma {\ Bigg|}\\\ hline
g (\ nu) =a\ nu^2 &\ fantasma {\ Bigg|}\\ hline
Q=\ dfrac {M!} {N! (M-N)!} Q^n &\ phantom {\ Bigg|}\\\ hline
\ Theta=\ dfrac {q\ exp\ bigg (\ dfrac {\ mu_0} {kT}\ bigg) p} {1+q\ exp\ bigg (\ dfrac {\ mu_0} {kT}\ bigg) p} &\ phantom {\ Bigg|}\\
pline _a=P_A^0x_A &\ phantom {\ Bigg|}\\\ hline
\ dfrac {c\ omega} {kT} =-4 &\ phantom {\ Bigg|}\\\ hline
W=W_ {AA} N_ {AA} +W_ {BB} N_ {BB} +W_ {AB} N_ {AB} &\ phantom {\ Bigg|}\\\ hline
N_ {AB}\ cong\ dfrac {N_A c N_B} {N_A+N_B} &\ phantom {\ Bigg|}\\\ hline\ end {array}\]
- Ley de Raoult
- Sólido Debye
- Punto Crítico
- Adsorción ideal
- Isoterma de Langmuir
- Bragg-Williams
- Función de partición para traducción de superficie
- Solución concentrada
- Fluctuación
- Coeficiente virial
- Sólido de Einstein
Problema al Tratar el Segundo Coeficiente Virial
56. La ecuación de estado de Van der Waals es
\[\left(p+\bigg(\dfrac{N}{V}\bigg)^2a\right)(V-Nb)=NkT\]
resolver esta ecuación para\(p\), y luego obtener una expresión para\(\dfrac{pV}{NkT}\). Por último, ampliar\(\dfrac{pV}{NkT}\) en poderes\(\bigg(\dfrac{N}{V}\bigg)\) y obtener una expresión para el segundo coeficiente virial de este gas Van der Waals en términos de\(b\),\(a\), y\(T\).
Problema para hacerte pensar en la realización de simulaciones de Montecarlo y Dinámica Molecular
57. Responda brevemente a cada uno de los siguientes:
Para cuál de los siguientes sería más sabio usar la simulación de Montecarlo (MC) y para cuál debería usar dinámica molecular (MD)
a. Determinar la velocidad de difusión de\(CH_4\) en líquido\(Kr\).
b. Determinar la distribución radial de equilibrio de los\(Kr\) átomos\(CH_4\) en relación con la del ejemplo anterior
c. Determinar la distancia cuadrática media de extremo a extremo para una cadena hidrocarbonada floja en estado líquido
Supongamos que está llevando a cabo una simulación de Montecarlo que involucra 1000\(Ar\) átomos. Además, supongamos que los potenciales son aditivos por pares y que su computadora requiere aproximadamente 50 operaciones de punto flotante (FPO) (por ejemplo, multiplicar, agregar, dividir, etc.) para calcular el potencial de interacción entre cualquier par de átomos
d. Por cada mudanza de juicio M-C, ¿cuántos FPO se requieren? Suponiendo que tu computadora tenga una velocidad de 100 MFlops (es decir, 100 millones de FPO's por segundo), ¿cuánto tiempo tardarás en llevar a cabo 1,000,000 de movimientos M-C?
e. Si las fluctuaciones observadas en el cálculo de la pregunta d son demasiado grandes, y desea hacer un cálculo M-C más largo para reducir el “ruido” estadístico, ¿cuánto tiempo requerirá su nuevo cálculo si desea reducir el ruido a la mitad?
f. ¿Cuánto tiempo requeriría el cálculo de la pregunta d si se utilizara 1,000,000 de\(Ar\) átomos (con el mismo potencial y la misma computadora)?
g. Suponiendo que la evaluación de las fuerzas entre pares de\(Ar\) átomos (\(\partial V/\partial r\);) requiere aproximadamente el mismo número de FPO's (50) que para calcular el potencial del par, cuánto tiempo (en segundos) tardaría en llevar a cabo una simulación de dinámica molecular que involucra 1000\(Ar\) átomos usando un paso de tiempo (\(\Delta t\)) de 10 -15 seg y persistiendo por una duración de tiempo total de un nanosegundo (10 -9 seg) usando la computadora 100 mFlop?
h. ¿Cuánto tiempo tardaría un estudiante de doctorado de 10 -6 MFlop (es decir, 1 FPO por segundo) para hacer el cálculo en la parte d?
Problema para practicar el uso de funciones de partición
58. En este problema, calculará la constante de equilibrio de la unidad de presión\(K_p\) para el equilibrio
\[2Na \rightleftharpoons Na_2\]
en la fase gaseosa a una temperatura de 1000 K. Su respuesta final debe expresarse en unidades de atm -1. Al hacerlo, debe considerar los símbolos electrónicos de términos de\(Na\) y de\(Na_2\), y deberá utilizar los siguientes datos:
i. no\(Na\) tiene estados electrónicos excitados que deba considerar.
ii. \(\dfrac{\hbar^2}{8\pi^2Ik} = 0.221\)K para\(Na_2\)
iii. \(\dfrac{h\nu}{k} = 229\)K para\(Na_2\)
iv. \(1 \text{ atm} = 1.01 \times 10^6 \text{ dynes cm}^{-2}\)
v. La energía de disociación\(Na_2\) del\(v\) = 0 a la disociación es\(D_0 = 17.3 \text{ kcal mol}^{-1}\).
a. primero, escribir las expresiones para las funciones\(Na\) y\(Na_2\) partición mostrando sus contribuciones traslacionales, rotacionales, vibracionales y electrónicas.
b. A continuación, sustituya los datos y cómpule\(K_p\), y cambie las unidades a atm -1.
Problema al usar la teoría del estado de transición
59. Mirando hacia atrás a la\(NO+Cl_2\) reacción tratada usando la teoría del estado de transición en el Problema 47, supongamos que esta misma reacción (a través del estado de transición doblado) iba a ocurrir mientras que a los reactivos\(NO\) y\(Cl_2\) se adsorbieron a una superficie de la siguiente manera:
a. ambos\(NO\) y\(Cl_2\) se encuentran planos contra la superficie con sus dos átomos tocando la superficie.
b. ambos\(NO\) y\(Cl_2\) se mueven libremente a lo largo de la superficie (es decir, pueden trasladarse paralelos a la superficie).
c. ambos\(NO\) y\(Cl_2\) están fuertemente unidos a la superficie de una manera que hace que sus movimientos perpendiculares a la superficie se conviertan en vibraciones de alta frecuencia.
Ante esta información, y asumiendo nuevamente las siguientes estimaciones de orden de magnitud de
funciones de partición
\[q_t \sim 10^8, q_r \sim 10^2, q_v \sim 1\]
calcular la relación de las constantes de velocidad TS para esta reacción que se produce en el estado adsorbido superficial y en la fase gaseosa. Al hacerlo, puede suponer que la energía de activación y todas las propiedades del estado de transición son idénticas en el estado gaseoso y adsorbido, excepto que las especies TS están obligadas a quedar planas en la superficie tal como son\(NO\) y\(Cl_2\).
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)