
6.5: Modelado Molecular
- Page ID
- 80703
Las formas de las moléculas se pueden predecir mediante el recuento de estimaciones de interacciones estéricas y torsionales. Esta tarea se conoce como un cálculo de mecánica molecular. El conjunto de bases utilizado puede variar del que se da aquí, pero la idea es la misma: se aplica un conjunto limitado de información a las moléculas, no importa cuán grandes o complejas sean, para medir la energía relativa de diferentes conformaciones. Normalmente un cálculo va seguido de un cambio en un ángulo diedro en alguna parte, el cálculo se repite y así sucesivamente, de manera que eventualmente se encuentra la conformación más estable.
Un cálculo como este podría llevar mucho tiempo. Incluso en una computadora, suele haber una necesidad de acelerar las cosas. Un atajo común para emplea el siguiente enfoque. Si un cambio en el ángulo diedro resulta en una disminución de la energía, el cálculo continúa con un nuevo cambio en ese ángulo diedro. Eventualmente, si los cambios conducen a un aumento de la energía, el programa asume que ya se ha pasado la energía más baja y vuelve a retroceder a ese punto. Luego puede probar otro diedro usando el mismo método. De esta manera la computadora no pierde tiempo en cálculos que involucran conformaciones eclipsadas de alta energía o con problemas estéricos. Como resultado, un cálculo de mecánica molecular generalmente puede estimar el conformador de energía más baja en pocos segundos.
La limitación de ese enfoque se puede ver en una gráfica genérica de energía vs. cambio conformacional en la que hay más de un conformador de baja energía. ¿Cómo determina el cálculo que ha encontrado el conformador de energía más bajo absoluto, y no solo una caída en la superficie de energía?
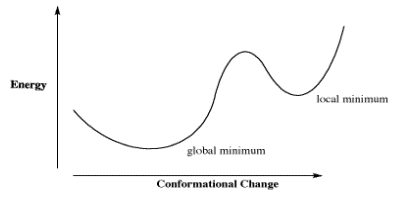
Por ejemplo, en una gráfica de la energía relativa del butano vs su ángulo diedro alrededor de C2-C3. La conformación gauche ocupa un “mínimo de energía local”, lo que significa que la energía es más baja aquí que cuando el ángulo diedro es mayor o menor, ya que en cualquier dirección la molécula llegaría a una conformación eclipsada con mayor energía. En el enfoque descrito anteriormente, típicamente el enfoque predeterminado en la mayoría de los cálculos, la computadora podría quedar atascada en la conformación gauche, porque un cambio en cualquier dirección da como resultado un aumento en la energía. No obstante, si la computadora simplemente empujara más allá de esos máximos de energía entonces llegaría a la anti conformación, que es el “mínimo de energía global”, con la energía más baja de todas.
- Tenga cuidado con los mínimos locales cuando busque el conformador más estable en una computadora.
- Por lo general, se puede decir si la computadora tiene la respuesta correcta en función de su comprensión de los esterics.
El análisis conformacional completo a veces requiere algún método de “conducción de ángulo diedro”. En este tipo de enfoque, el usuario del software puede decirle a la computadora que calcule energías un número dado de veces alrededor de un ángulo diedro dado; por ejemplo, tal vez se haría un cálculo cada sesenta grados para el diedro alrededor del enlace C2-C3 en butano, o tal vez cada treinta grados. De esa manera, la computadora seguramente encontrará el mínimo global para un ángulo diedro particular.
Se pueden requerir métodos más avanzados para llegar a estructuras correctas de baja energía para moléculas más complicadas. Por ejemplo, los enlaces de hidrógeno son claramente un factor importante que controla la estructura de moléculas como el ADN y las proteínas. Solo hemos analizado las cepas estéricas y torsionales porque son ubicuas en la mecánica molecular, pero diferentes conjuntos de bases, y diferentes tipos de cálculos, podrían tener en cuenta interacciones adicionales como los enlaces de hidrógeno u otros fenómenos electrónicos que ayudarían a establecer correctamente el formas de moléculas más complejas.