8.9: Destilación
- Page ID
- 70916

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Asegúrese de comprender a fondo las siguientes ideas esenciales:
- Esboce un diagrama de punto de ebullición típico para una solución líquida binaria y utilícelo para mostrar cómo funciona una destilación simple de una etapa.
- Explicar el papel de la regla de palanca en la destilación fraccionada
- Describir el propósito y función de una columna de fraccionamiento
- Dibuje diagramas de punto de ebullición para azeótropos de alto y bajo punto de ebullición
- Describir el papel de la destilación en la refinación de petróleo crudo y explicar, de manera muy general, cómo se utiliza el procesamiento adicional para aumentar el rendimiento del combustible para motores de gasolina.
La destilación es un proceso mediante el cual una mezcla de líquidos que tienen diferentes presiones de vapor se separa en sus componentes. Al principio se podría pensar que esto sería bastante sencillo: si se tiene una solución que consiste en líquido A que hierve a 50°C y líquido B con un punto de ebullición de 90°C, todo lo que sería necesario sería calentar la mezcla a alguna temperatura entre estos dos valores; esto herviría toda la A (cuyo vapor podría entonces condensarse de nuevo en líquido puro A), dejando el líquido B puro en la olla. Pero eso pasa por alto ese hecho de que estos líquidos tendrán presiones de vapor sustanciales a todas las temperaturas, no sólo en sus puntos de ebullición.
Diagramas de Fase de Presión de Vapor
Para comprender completamente la destilación, consideraremos una mezcla líquida binaria ideal de\(\ce{A}\) y\(\ce{B}\). Si la fracción molar de\(A\) en la mezcla es\(\chi_A\), entonces por la definición de fracción molar, la de\(B\) es
\[\chi_B = 1 – \chi_A\]
Dado que la destilación depende de las diferentes presiones de vapor de los componentes a separar, primero consideremos las gráficas de presión de vapor vs. composición (Figura\(\PageIndex{1}\)) para una mezcla hipotética a alguna temperatura arbitraria a la que puedan existir ambas fases líquida y gaseosa, dependiendo de la presión total.
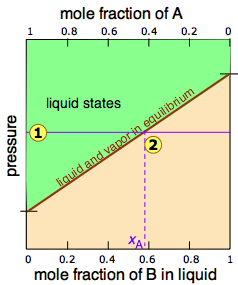
En la Figura\(\PageIndex{2}\), todos los estados del sistema (es decir, combinaciones de presión y composición) en los que la solución existe únicamente como líquido están sombreados en verde. Dado que los líquidos son más estables a presiones más altas, estos estados ocupan la parte superior del diagrama. A cualquier presión de vapor total dada como at , la composición del vapor en equilibrio con el líquido (designada por\(x_A\)) corresponde a la intersección con la línea de equilibrio diagonal at
, la composición del vapor en equilibrio con el líquido (designada por\(x_A\)) corresponde a la intersección con la línea de equilibrio diagonal at . La línea diagonal es solo una expresión de la linealidad entre la presión de vapor y la composición según la ley de Raoult.
. La línea diagonal es solo una expresión de la linealidad entre la presión de vapor y la composición según la ley de Raoult.
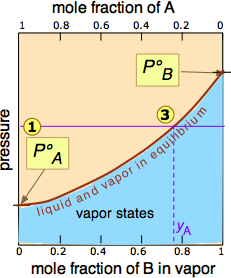
 de la línea de presión con la curva de equilibrio define las fracciones molares de\(\ce{A}\) y\(\ce{B}\) presentes en el vapor. (Obsérvese que las fracciones molares de gases, de las que estamos tratando aquí, están representadas convencionalmente por y, de ahí y A e y B). La curvatura de la línea de equilibrio surge de la necesidad de combinar la ley de Raoult con la ley de Dalton de presiones parciales que se aplica a las mezclas gaseosas.
de la línea de presión con la curva de equilibrio define las fracciones molares de\(\ce{A}\) y\(\ce{B}\) presentes en el vapor. (Obsérvese que las fracciones molares de gases, de las que estamos tratando aquí, están representadas convencionalmente por y, de ahí y A e y B). La curvatura de la línea de equilibrio surge de la necesidad de combinar la ley de Raoult con la ley de Dalton de presiones parciales que se aplica a las mezclas gaseosas.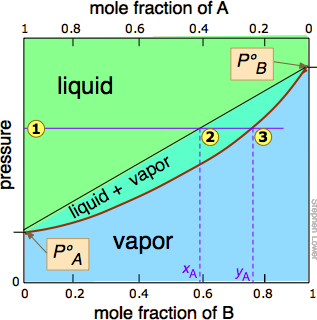
Las dos líneas de equilibrio líquido-vapor (una curvada, la otra recta) encierran ahora un área en la que pueden coexistir líquido y vapor; fuera de esta región, la mezcla consistirá enteramente en líquido o vapor. A esta presión particular, la intercepción con el límite superior de la región bifásica da las fracciones molares de A y B en la fase líquida, mientras que la intersección con el límite inferior da las fracciones molares de los dos componentes en el vapor.
Tómese un momento para estudiar Figura\(\PageIndex{5}\) y confirmar que
- debido a que ambas intercepciones ocurren en líneas de equilibrio, describen las composiciones del líquido y vapor que pueden existir simultáneamente;
- las composiciones del vapor y el líquido no son las mismas;
- en el vapor, la fracción molar de\(\ce{B}\) (el componente más volátil de la solución) es mayor que la del líquido;
- en el líquido, la fracción molar de\(\ce{A}\) (el componente menos volátil) es menor que la del vapor.
El vapor en equilibrio con una solución de dos o más líquidos es siempre más rico en el componente más volátil.
Diagramas de fases de temperatura vs. composición (diagramas de punto de ebullición
La regla mostrada anteriormente sugiere que si calentamos una mezcla suficientemente para llevar su presión de vapor total a la región bifásica, tendremos un medio para separar la mezcla en dos porciones las cuales serán enriquecidas en los componentes más volátiles y menos volátiles respectivamente. Este es el principio en el que se basa la destilación. Pero, ¿qué temperatura se requiere para lograr esto? Nuevamente, le ahorraremos los detalles matemáticos, pero es posible construir una gráfica similar a la Figura\(\PageIndex{4}\) excepto que el eje vertical representa la temperatura más que la presión. Este tipo de trama se llama diagrama de punto de ebullición.
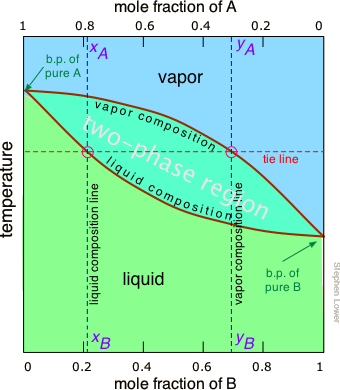
Propiedades importantes de los diagramas de punto de ebullición
Algunas cosas importantes a entender sobre la Figura\(\PageIndex{6}\):
- La forma de la región bifásica es biconvexa, a diferencia de la forma semiconvexa de la gráfica presión-composición.
- La pendiente de la región bifásica es opuesta a la que vimos en la parcela anterior, y las áreas correspondientes a las regiones monofásicas se invierten. Esto simplemente refleja el hecho de que los líquidos que tienen una mayor presión de vapor hierven a temperaturas más bajas, y viceversa.
- La línea horizontal que define la temperatura se llama línea de unión. Sus intercepciones con las dos curvas de equilibrio especifican la composición del líquido y vapor en equilibrio con la mezcla a la temperatura dada.
- La línea de composición de vapor también se conoce como la línea de punto de rocío, la temperatura a la que la condensación comienza al enfriarse.
- La línea de composición líquida también se llama línea de punto de burbuja, la temperatura a la que comienza la ebullición al calentarse.
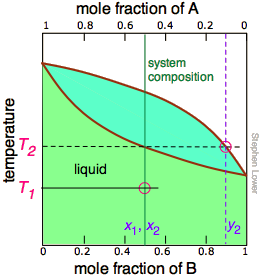
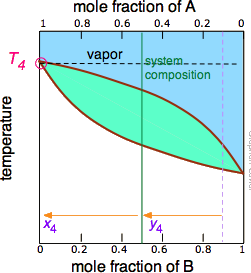
La línea de unión que se muestra en la Figura\(\PageIndex{6}\) es para una temperatura particular. Pero cuando calentamos un líquido a su punto de ebullición, la composición cambiará a medida que el componente más volátil (\(\ce{B}\)en estos ejemplos) se elimine selectivamente como vapor. El líquido restante se enriquecerá en el componente menos volátil, y su punto de ebullición en consecuencia aumentará. Para entender más a fondo este proceso, consideremos la situación en varios puntos durante la destilación de una solución equimolar de\(\ce{A}\) y\(\ce{B}\).
 |
 |
 |
|
Figura\(\PageIndex{5A}\): Comenzamos con el líquido en T 1, por debajo de su punto de ebullición. Cuando la temperatura sube a T 2, comienza la ebullición y el primer vapor (y así la primera gota de condensado) tendrá la composición y 2. |
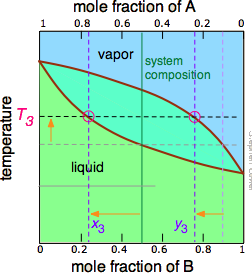
Figura\(\PageIndex{5B}\): A medida que se hierve el componente B más volátil, las composiciones líquida y vapor/condensado se desplazan hacia la izquierda (flechas naranjas). | Figura\(\PageIndex{5C}\): A T 4, el último rastro de líquido desaparece. El sistema ahora es completamente vapor, de composición y 4. |
Observe que la línea de composición del sistema verde vertical permanece en la misma ubicación en las tres parcelas porque el “sistema” se define como consistente tanto en el líquido en la “olla” como en el recipiente receptor que se condensó a partir del vapor. Las ideas principales que debes quitar de esto son que
- la destilación nunca puede separar completamente dos líquidos volátiles;
- la composición del vapor y por lo tanto del destilado condensado cambia continuamente a medida que se forma cada gota, comenzando en y 2 y terminando en y 4 en este ejemplo;
- si el líquido está completamente hervido, la composición del destilado será la misma que la de la solución original.
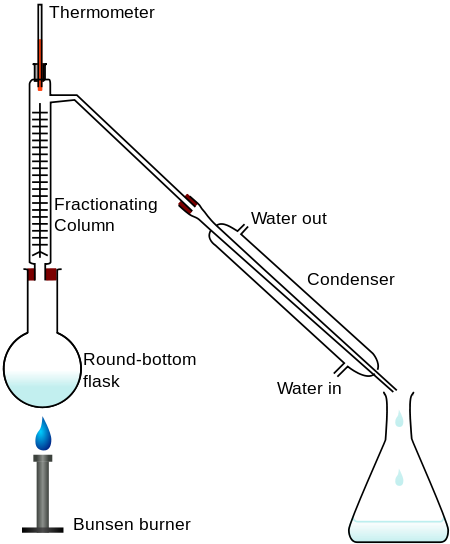
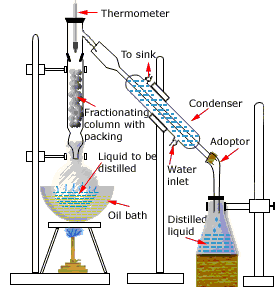
Configuración de destilación de laboratorio
Aquí se muestra el aparato utilizado para una simple destilación por lotes de laboratorio. El propósito del termómetro es seguir el progreso de la destilación; como regla general aproximada, la destilación debe detenerse cuando la temperatura suba aproximadamente a medio camino entre los puntos de ebullición de los dos líquidos puros, que deben estar separados por lo menos 20-30 C° (si están más cerca, entonces la destilación fraccionada, descrita a continuación, se vuelve necesaria).
Destilación Fraccional
Aunque la destilación nunca puede lograr la separación completa de los líquidos volátiles, en principio se puede llevar a cabo de tal manera que se pueda lograr cualquier grado de separación deseado si la solución se comporta de manera ideal y uno está dispuesto a ir a la molestia. El procedimiento general es destilar solo una fracción del líquido, cuanto más pequeño mejor. El condensado, ahora enriquecido en el componente más volátil, es luego recogido y redestilado (nuevamente, solo una pequeña fracción), obteniendo así un condensado aún más enriquecido en el componente más volátil. Si repetimos esta secuencia muchas veces, eventualmente podremos obtener muestras casi puras, si diminutas, de los dos componentes.
Pero como esto difícilmente sería práctico, hay una mejor manera. Para entenderlo, es necesario conocer la regla de palanca, que proporciona una forma sencilla de determinar las cantidades relativas (no solo las composiciones) de dos fases en equilibrio. La regla de la palanca se deriva fácilmente de las leyes de Raoult y Dalton, pero simplemente la ilustraremos gráficamente (Figura\(\PageIndex{7}\)). La gráfica muestra el diagrama de punto de ebullición de una mezcla binaria simple de composición. A la temperatura correspondiente a la línea de unión, la composición del líquido corresponde a y la del vapor a.
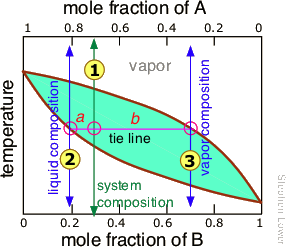
Entonces ahora para la regla de palanca: las cantidades relativas del líquido y el vapor que identificamos anteriormente están dadas por las longitudes de los segmentos de línea de unión etiquetados a y b. Así, en este ejemplo particular, en el que b es aproximadamente cuatro veces más largo que a, podemos decir que la relación molar de vapor (de composición) a líquido (composición
) es de 4.
Pasos en la destilación fraccionada
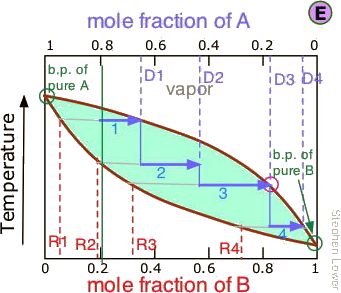
No es práctico llevar a cabo un número casi infinito de etapas de destilación para obtener cantidades casi infinitesimales de los dos líquidos puros que deseamos separar. Entonces, en lugar de recoger cada gota de condensado y volver a destilarla, destilaremos la mitad de la mezcla en cada paso. Supongamos que desea separar una mezcla líquida compuesta por 20 moles-% B y 80 mole-% A, siendo A la más volátil.
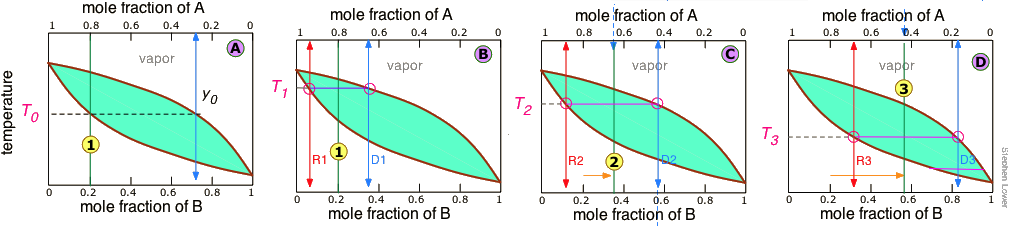
 A medida que calentamos la mezcla cuya composición general se indica por, el primer vapor se forma en T 0 y tiene la composición y 0, que se encuentra extendiendo la línea discontinua horizontal hasta que cumpla con la curva de vapor. Este vapor está claramente enriquecido en B; si se condensa, el líquido resultante tendrá una fracción molar x B acercándose a la de A en el líquido original. Pero esta es sólo la primera gota, ¡no queremos detenernos ahí!
A medida que calentamos la mezcla cuya composición general se indica por, el primer vapor se forma en T 0 y tiene la composición y 0, que se encuentra extendiendo la línea discontinua horizontal hasta que cumpla con la curva de vapor. Este vapor está claramente enriquecido en B; si se condensa, el líquido resultante tendrá una fracción molar x B acercándose a la de A en el líquido original. Pero esta es sólo la primera gota, ¡no queremos detenernos ahí!
 A medida que el líquido continúa hirviendo, la temperatura de ebullición aumenta. Cuando llegue a T 1, habremos evaporado la mitad del líquido. En este punto, la composición del “sistema” (líquido más vapor) sigue siendo la misma (), pero ahora se divide por igual entre el líquido, que llamamos “residuo” R1, y el vapor condensado, el destilado D 1.
A medida que el líquido continúa hirviendo, la temperatura de ebullición aumenta. Cuando llegue a T 1, habremos evaporado la mitad del líquido. En este punto, la composición del “sistema” (líquido más vapor) sigue siendo la misma (), pero ahora se divide por igual entre el líquido, que llamamos “residuo” R1, y el vapor condensado, el destilado D 1.
¿Cómo sabemos que está dividido por partes iguales? Hemos escogido T 1 para que la línea de unión esté centrada en la concentración del sistema, por lo que por la regla de la palanca, R1 y D 1 contienen números iguales de moles.
 Tomamos ahora el líquido condensado D 1 que tiene la composición, y destilamos la mitad del mismo, obteniendo destilado de composición D 2.
Tomamos ahora el líquido condensado D 1 que tiene la composición, y destilamos la mitad del mismo, obteniendo destilado de composición D 2.
 .. y luego llevar a cabo otra destilación, esta vez usando D 3 como nuestra materia prima.
.. y luego llevar a cabo otra destilación, esta vez usando D 3 como nuestra materia prima.
 Nuestro fraccionamiento de cuatro etapas ha enriquecido al soluto más volátil de 20 a ligeramente más del 80 por ciento en moles en D 4. El componente A menos volátil está más concentrado en R1. R 2 a R 4 se tiran a la basura (¡pero no por el fregadero, por favor!)
Nuestro fraccionamiento de cuatro etapas ha enriquecido al soluto más volátil de 20 a ligeramente más del 80 por ciento en moles en D 4. El componente A menos volátil está más concentrado en R1. R 2 a R 4 se tiran a la basura (¡pero no por el fregadero, por favor!)
Esto puede ser suficiente para algunos propósitos, pero tal vez deseemos hacerlo mucho mejor, usando quizás 1000 etapas en lugar de solo 4. ¿Qué podría ser más tedioso?
Fraccionamiento con reflujo
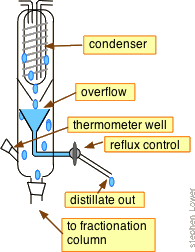
¡No te preocupes! Las múltiples destilaciones sucesivas se pueden llevar a cabo “virtualmente” insertando una columna de fraccionamiento entre el matraz de ebullición y el condensador.
Estas columnas están hechas con indentaciones o están llenas de materiales que proporcionan una gran superficie que se extiende a través del gradiente de temperatura vertical (mayor temperatura cerca del fondo, menor temperatura en la parte superior). La idea es que los vapores calientes se condensen a varios niveles en la columna y el líquido resultante gotee hacia abajo (refluye) a un nivel inferior donde se vaporiza, lo que corresponde aproximadamente a una redestilación.

Las columnas Vigreux que tienen múltiples hendiduras son ampliamente utilizadas (arriba a la derecha). Se pueden hacer columnas simples llenando un tubo de vidrio con cuentas, tubos cortos de vidrio o incluso estropajos tipo cocina de acero inoxidable. Los más elaborados tienen cintas de acero giratorias.
Eficiencia de separación: placas teóricas
El funcionamiento de las columnas de fraccionamiento se puede entender mejor por referencia a una columna con tapa de burbujas. El que aquí se muestra consta de cuatro secciones, o “placas” a través de las cuales los vapores calientes suben y burbujean a través de charcas de condensado que se recogen en cada placa. El contacto íntimo entre vapor y líquido promueve el equilibrio y la redestilación a temperaturas sucesivamente más altas en cada placa superior de la columna. A diferencia del caso de la destilación fraccionada escalonada que discutimos anteriormente, ninguno de los residuos intermedios es desechado; simplemente gotean de nuevo al bote donde comienza nuevamente su viaje de fraccionamiento, conduciendo siempre a una mayor concentración del componente menos volátil en el líquido restante. Al mismo tiempo, el vapor que emerge de la placa superior (5) proporciona un flujo continuo de condensado enriquecido en volátiles, aunque en cantidades decrecientes a medida que se agota en la olla de ebullición.
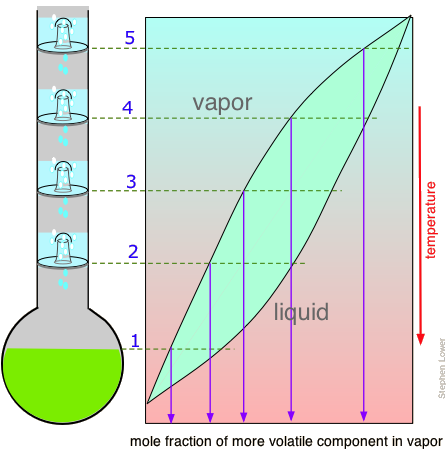
Si se logra un equilibrio completo entre el líquido y el vapor en cada etapa, entonces podemos describir el sistema ilustrado anteriormente como proporcionar “cinco placas teóricas” de separación (recuerde que el bote representa la primera placa teórica). El equilibrio en cada etapa requiere una condición de estado estacionario en la que la cantidad de vapor que se mueve hacia arriba en cada etapa es igual a la cantidad de líquido que drena hacia abajo, es decir, la columna debe estar operando en reflujo total, sin eliminación neta de destilado. Por lo que cualquier proceso de destilación real se operará a una relación de reflujo que proporcione una separación óptima en un periodo de tiempo razonable.
Se dice que algunos de los dispositivos de tipo laboratorio más avanzados (como algunas columnas de banda giratoria de acero) ofrecen hasta alrededor de 200 placas teóricas de potencia de separación.
Azeótropos: los límites de la destilación
Los diagramas de punto de ebullición presentados en el apartado anterior se aplican a soluciones que se comportan de manera razonablemente ideal, es decir, a soluciones que no se apartan demasiado de la ley de Raoult. Como explicamos anteriormente, las mezclas de líquidos cuyas interacciones intermoleculares son muy diferentes no se comportan idealmente, y pueden ser imposibles de separar por destilación ordinaria. La razón de esto es que bajo ciertas condiciones, las composiciones del líquido y del vapor en equilibrio con él se vuelven idénticas, impidiendo cualquier separación adicional. Estos puntos de cruce aparecen como “torceduras” en los diagramas de punto de ebullición.
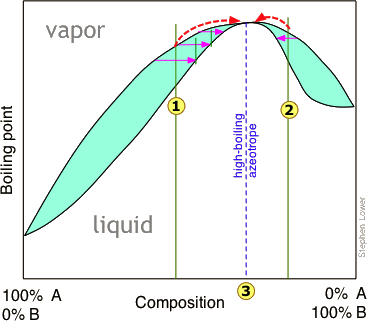
Azeótropos de alto y bajo punto de ebullición
Así, en este diagrama de punto de ebullición para una mezcla que presenta una desviación positiva de la ley de Raoult, sucesivos fraccionamientos de mezclas corresponden o acercan la destilación a la composición azeotrópica indicada por la línea vertical discontinua. Una vez que se alcanza este punto, la destilación adicional simplemente produce más del mismo azeótropo de “alto punto de ebullición”.
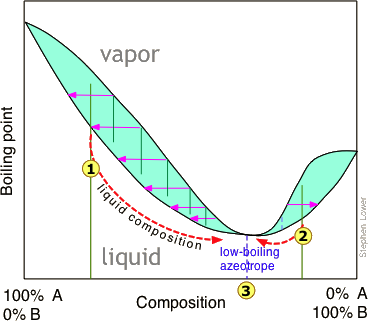
La destilación de una mezcla que tiene una desviación negativa de la ley de Raoult conduce a un estancamiento similar, en este caso produciendo un azeótropo de “bajo punto de ebullición”. Los azeótropos de alto y bajo punto de ebullición se conocen comúnmente como mezclas de ebullición constante, y son más comunes de lo que la mayoría de la gente piensa.
“Rompiendo” un azeótropo
Hay cuatro formas generales de tratar los azeótropos. Los dos primeros de estos se conocen colectivamente como destilación azeotrópica.
- La adición de una tercera sustancia que altera las atracciones intermoleculares es el truco más común. El inconveniente es que generalmente se necesita otro procedimiento para eliminar esta otra sustancia.
- La destilación por oscilación de presión aprovecha que los diagramas de punto de ebullición (T, X) son rebanadas bidimensionales de un diagrama (T, X, P) en el que la presión es la tercera variable. Esto significa que la composición azeotrópica depende de la presión, por lo que la destilación a alguna presión distinta de 1 atm puede permitir que uno “salte” el azeótropo.
- Uso de un tamiz molecular — un material poroso que absorbe selectivamente uno de los líquidos, más comúnmente agua cuando este último está presente en una concentración baja.
- Ríndete. A menudo sucede que la composición azeotrópica es lo suficientemente útil como para que normalmente no valga la pena la molestia de obtener un producto más puro. Esto explica las concentraciones de muchos químicos comerciales como los ácidos minerales.
| mezcla | azeótropo |
|---|---|
| Etanol | 98%, alto, 78.1°C |
| Ácido clorhídrico | 20.2% alto, 108.6°C |
| Ácido fluorhídrico | 35.6%, 111.3°C |
| Ácido nítrico | 68%, 120.5°C |
| Ácido sulfúrico | 98.3%, 338°C |
Destilación de etanol
El etanol es uno de los principales productos químicos industriales y, por supuesto, es el componente esencial de las bebidas que han sido parte de la civilización a lo largo de la historia registrada. La mayor parte del etanol se produce por fermentación del almidón presente en los granos alimenticios, o de azúcares formados por la degradación enzimática de la celulosa. Debido a que el etanol es tóxico para los organismos cuyas enzimas median en el proceso de fermentación, la concentración de etanol en la mezcla fermentada generalmente se limita a aproximadamente 15%. Después se separa la fase líquida de la mezcla y se destila.
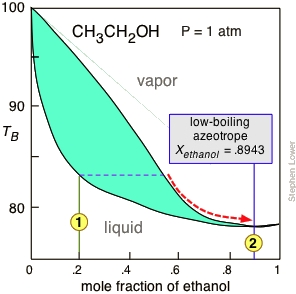 Figura\(\PageIndex{8}\): El punto de ebullición normal del etanol es de 78.4°C, pero su formación de un azeótropo de bajo punto de ebullición con agua limita la concentración máxima que se puede obtener b y ordinaria destilación a 95.6 volumen-% (89.5 mole-%). Esta concentración es adecuada para muchas aplicaciones, para las cuales el producto se vende comúnmente como "95% etano o l”.
Figura\(\PageIndex{8}\): El punto de ebullición normal del etanol es de 78.4°C, pero su formación de un azeótropo de bajo punto de ebullición con agua limita la concentración máxima que se puede obtener b y ordinaria destilación a 95.6 volumen-% (89.5 mole-%). Esta concentración es adecuada para muchas aplicaciones, para las cuales el producto se vende comúnmente como "95% etano o l”.
Para aplicaciones que requieren etanol anhidro (“etanol absoluto “), el método más común es el uso de tamices moleculares a base de zeolita para absorber el agua restante. La adición de benceno puede romper el azeótropo, y este fue el método de producción más común en años anteriores. Para ciertos usos críticos donde se requiere el etanol más puro, se sintetiza directamente a partir del etileno.
Métodos especiales de destilación
Aquí discuta brevemente dos métodos de destilación que es probable que los estudiantes encuentren en cursos de laboratorio orgánico más avanzados.
Destilación al vacío: Muchas sustancias orgánicas se vuelven inestables a altas temperaturas, tendiendo a descomponerse, polimerizarse o reaccionar con otras sustancias a temperaturas alrededor de 200° C o superiores. Un líquido hervirá cuando su presión de vapor llegue a ser igual a la presión del gas por encima de él, que normalmente es la de la atmósfera. Si se reduce esta presión, la ebullición puede tener lugar a una temperatura más baja. (Incluso el agua pura hervirá a temperatura ambiente bajo vacío parcial). “Destilación al vacío” es, por supuesto, un nombre inapropiado; un término más preciso sería “destilación a presión reducida”. La destilación al vacío se lleva a cabo muy comúnmente en el laboratorio y será familiar para los estudiantes que toman cursos de laboratorio orgánico más avanzados. También se emplea a veces a gran escala industrial.
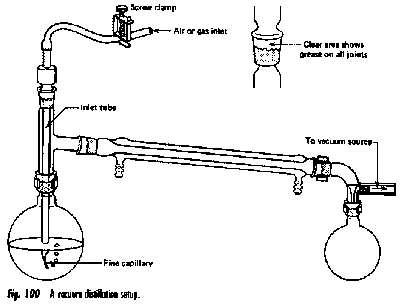

La configuración de destilación al vacío es similar a la empleada en la destilación ordinaria, con algunas adiciones:
- La línea de vacío está conectada al adaptador doblado encima del matraz receptor.
- Para evitar la ebullición desigual y el sobrecalentamiento (“golpes”), el matraz de ebullición suele estar provisto de un capilar fino (“ebulador”) a través del cual una fuga de aire produce burbujas que nuclean el líquido hirviendo.
- El vacío generalmente es suministrado por una bomba mecánica, o menos comúnmente por un aspirador de agua o una línea de “vacío doméstico”.
- El matraz de ebullición se calienta preferiblemente mediante un baño de agua o vapor, lo que proporciona una transferencia de calor más eficiente al matraz y evita el sobrecalentamiento localizado. Antes de alrededor de 1960, las llamas abiertas se usaban comúnmente en los laboratorios estudiantiles, dando como resultado incendios ocasionales que animaban la tarde, pero restaban valor a las marcas de laboratorio del estudiante.
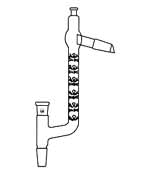
- Un cabezal de destilación tipo Claisen (abajo) proporciona un medio conveniente para acceder al matraz de ebullición para insertar un capilar de fuga de aire o introducir líquido adicional a través de un embudo separador. Esta cabeza Claisen-Vigreux incluye una columna de fraccionamiento.
Destilación a Vapor: Estrictamente hablando, este tema no pertenece a esta unidad, ya que la destilación al vapor se utiliza para separar líquidos inmiscibles en lugar de soluciones. Pero debido a que las mezclas líquidas inmiscibles no son tratadas en cursos elementales, aquí presentamos una breve descripción de la destilación al vapor en beneficio de los estudiantes que pueden encontrarla en un curso de laboratorio orgánico. Una mezcla de líquidos inmiscibles hervirá cuando sus presiones de vapor combinadas alcancen la presión atmosférica. Esta presión de vapor combinada es solo la suma de las presiones de vapor de cada líquido individualmente, y es independiente de las cantidades de cada fase presente.
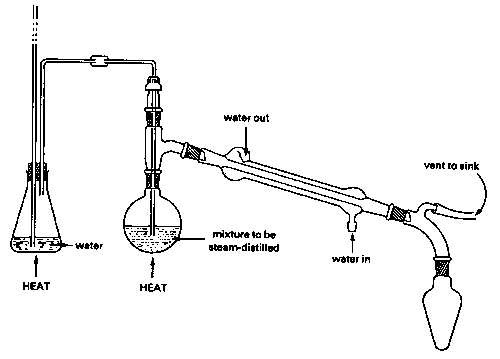
Debido a que el agua hierve a 100° C, se garantiza que una mezcla de agua y un líquido inmiscible (un “aceite”), incluso uno que tenga un punto de ebullición alto, hierva por debajo de los 100°, por lo que este método es especialmente valioso para separar líquidos de alto punto de ebullición de mezclas que contienen impurezas no volátiles. Por supuesto, la mezcla agua-aceite en el matraz receptor debe separarse por sí misma, pero esto generalmente se logra fácilmente por medio de un embudo separador ya que sus densidades son normalmente diferentes.
Hay una captura, sin embargo: cuanto menor es la presión de vapor del aceite, mayor es la cantidad de agua que codestila con él. Esta es la razón para usar vapor: proporciona una fuente de agua capaz de restaurar continuamente lo que se pierde del matraz hirviendo. La destilación al vapor de una mezcla de agua-aceite sin la introducción de vapor adicional también funcionará, y en realidad se usa para algunos propósitos especiales, pero el rendimiento del producto será muy limitado. La destilación al vapor es ampliamente utilizada en industrias como la refinación de petróleo (donde a menudo se llama “extracción por vapor”) y en la industria de aromas y perfumes para el aislamiento de aceites esenciales
El término aceite esencial se refiere a los aromas (“esencias”) de estos líquidos orgánicos [en su mayoría simples] que ocurren naturalmente en las plantas, de los cuales se aíslan por destilación al vapor o extracción por solvente. La destilación al vapor fue inventada en el siglo XIII por Ibn al-Baiter, uno de los más grandes científicos y médicos de la Edad de Oro islámica en Andalucía.
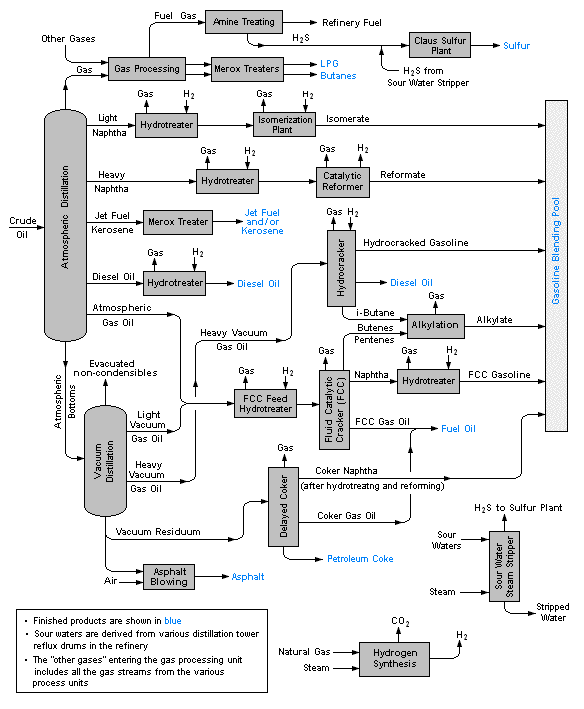
Destilación a escala industrial y fraccionamiento de petróleo
La destilación es una de las principales “operaciones unitarias” de las industrias de procesos químicos, especialmente las relacionadas con la refinación de petróleo y biocombustibles, la separación de aire líquido y la elaboración de cerveza. Las destilaciones de laboratorio son típicamente operaciones discontinuas y emplean columnas de fraccionamiento relativamente simples para obtener un producto puro. Por el contrario, las destilaciones industriales se diseñan con mayor frecuencia para producir mezclas que tienen un rango de ebullición deseado en lugar de productos puros.
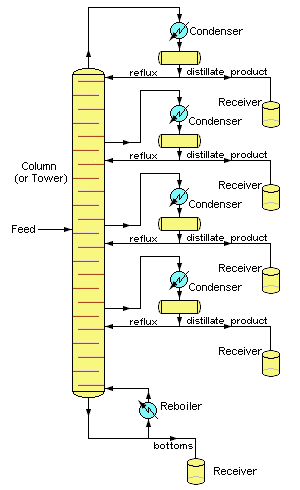
Las operaciones industriales suelen emplear columnas de fraccionamiento con tapa burbujeante (raramente vistas en laboratorios), aunque a veces se utilizan columnas empaquetadas. Quizás la característica más distintiva de las destilaciones industriales a gran escala es que generalmente operan de forma continua en la que la mezcla cruda precalentada se precalienta en un horno y se alimenta a la columna de fraccionamiento en algún punto intermedio. Una unidad de calderín mantiene la temperatura del fondo a un valor constante. Los componentes de mayor punto de ebullición se mueven entonces hacia abajo a un nivel en el que se vaporizan, mientras que el material más ligero (punto de ebullición más bajo) se mueve hacia arriba para condensarse en un punto apropiado.
El petróleo es una mezcla compleja de muchos tipos de moléculas orgánicas, en su mayoría hidrocarburos, que se formaron por los efectos del calor y la presión sobre los materiales vegetales (principalmente algas) que crecieron en regiones que los movimientos tectónicos de la tierra enterraron durante periodos de millones de años. Esta mezcla de líquidos y gases migra hacia arriba a través de roca porosa hasta quedar atrapada por una capa impermeable de roca sedimentaria. La composición molecular del petróleo crudo (la fracción líquida del petróleo) es altamente variable, aunque su composición elemental general generalmente refleja la de plantas típicas.
| elemento | carbono | hidrógeno | nitrógeno | oxígeno | azufre | metales |
|---|---|---|---|---|---|---|
| cantidad | 83-87% | 10-14% | 0.1-2% | 0.1-1.5% | 0.5-6% |
Los principales constituyentes moleculares del petróleo crudo son
- alcanos: También conocidas como parafinas, estas son moléculas saturadas de cadena lineal o ramificada que tienen la fórmula general C n H 2 n +2 en la que n está mayormente entre 5 y 40.
- alifático insaturado: Moléculas de cadena lineal o ramificada que contienen uno o más dobles o triples enlaces (alquenos o alquinos).
- Cicloalcanos:También conocidos como naftenos estos son hidrocarburos saturados C n H 2 n que contienen una o más estructuras de anillo.
- Hidrocarburos aromáticos:Estos contienen uno o más anillos de benceno condensados C n H n, a menudo con cadenas laterales de hidrocarburos.
La palabra gasolina es anterior a su uso como combustible para motores; primero se utilizó como medicina tópica para librar a las personas de los piojos de la cabeza, y para eliminar manchas de grasa y manchas de la ropa. El primer paso importante de la refinación es fraccionar el petróleo crudo en varios rangos de ebullición.
| rango de ebullición | nombre de la fracción | procesamiento adicional |
|---|---|---|
| butano y propano | procesamiento de gas | |
| 30 - 210° | gasolina en línea recta | mezcla en gasolina de motor |
| 100 - 200° | nafta | reformar en componentes de gasolina |
| 150 - 250° | queroseno | mezcla de combustible para aviones |
| 160 -400° | gasóleo ligero | mezcla de combustible destilado en diesel o fueloil |
| 315 - 540° | gasóleo pesado | craqueo catalítico: las moléculas grandes se descomponen en otras más pequeñas y se reciclan |
| >450° | asfaltos, fondos | puede ser destilado al vacío en más fracciones |
Procesamiento y mezcla adicionales
Alrededor del 16% del petróleo crudo se desvía a la industria petroquímica donde se utiliza para fabricar etileno y otras materias primas para plásticos y productos similares. Debido a que la fracción de gasolina de ciclo directo es inadecuada para satisfacer la demanda, algunas de las fracciones más ligeras se someten a reformas y las más pesadas se agrietan y se reciclan en la corriente de gasolina. Estos procesos requieren una gran cantidad de reciclaje y mezcla, en los que se debe construir una cantidad considerable de flexibilidad para satisfacer las necesidades estacionales (más gasolinas volátiles y fuelóleo para calefacción en invierno, más volúmenes totales de gasolina en verano).