
17.1: Tasas de reacciones y leyes de tarifas
- Page ID
- 70604

Asegúrese de comprender a fondo las siguientes ideas esenciales:
- Describir los roles contrastantes de la termodinámica y la cinética en la comprensión del cambio químico.
- Dada una ecuación neta equilibrada, escribir una expresión para la velocidad de una reacción.
- Haga un boceto de una curva que muestre cómo la velocidad instantánea de una reacción podría cambiar con el tiempo.
- Determinar el orden de una reacción de la forma A → B + C a partir de datos experimentales para las concentraciones de sus productos en tiempos sucesivos.
- Describir la velocidad inicial y los métodos de aislamiento para determinar los órdenes de los reactivos individuales en una reacción que involucra múltiples reactivos.
- Explicar la diferencia entre las leyes de tasas diferenciales e integrales.
- Dibuje una gráfica que muestre cómo la concentración de un componente ([A] o ln [A]) que sigue la cinética de primer orden cambiará con el tiempo. Indicar cómo la magnitud de la constante de velocidad afecta a esta parcela
- Definir la vida media de una reacción.
- Dada la vida media para una reacción de primer orden A → productos junto con el valor inicial de [A] o, encontrar [A] t en un momento posterior un número entero de semividas después.
- Describir las condiciones bajo las cuales una reacción puede parecer tener un orden de cero.
El cambio químico es guiado e impulsado por la energética, pero la ruta real que toma y la velocidad con la que ocurre es objeto de “dinámica”. La dinámica se divide en dos áreas generales: la cinética, que trata de la tasa de cambio y es el tema de esta lección. La mecánica, introducida en una lección posterior, es una exploración de la “hoja de ruta” que vincula los reactivos con los productos.
Los aspectos energéticos del cambio se rigen por las leyes de la termodinámica (la parte “dinámica” de esta palabra está relacionada con los orígenes históricos del campo y no forma parte de la dinámica en el sentido de estas lecciones).
Energética + dinámica = cambio químico
El cambio químico es impulsado por la tendencia de los átomos y moléculas a reorganizarse de manera que resulte en la máxima dispersión posible de la energía térmica en el mundo. La cantidad observable que mide esta difusión y distribución de energía es la energía libre del sistema. A medida que se produce un cambio químico, las cantidades de reactivos y productos cambian de una manera que conduce a una energía libre más negativa. Cuando la energía libre alcanza su valor mínimo posible, no hay más cambio neto y se dice que el sistema está en equilibrio.
La belleza de la termodinámica es que nos permite predecir indefectiblemente la dirección neta de una reacción y la composición del estado de equilibrio incluso sin realizar el experimento; las energías libres estándar de los reactivos y productos, que pueden medirse de manera independiente u obtenerse de tablas, son todo lo que necesitamos.
La mitad de la historia
La termodinámica señala el camino y lo hace posible... ¡pero no dice nada sobre cuánto tiempo tardará en llegar ahí!
La ecuación estequiométrica para la reacción no dice nada sobre su mecanismo. Por mecanismo, nos referimos, básicamente, a “quién hace qué a quién”. Piense en un mecanismo de reacción como algo que sucede en una “caja negra” que une los reactivos a los productos:

El funcionamiento interno de la caja negra normalmente se nos oculta, son altamente impredecibles y solo pueden inferirse por medios indirectos. Consideremos, por ejemplo, las reacciones de formación en fase gaseosa de los haluros de hidrógeno de los elementos.
La ecuación Equilibrada no dice nada sobre el Mecanismo de una Reacción
La termodinámica de estas reacciones son todas similares (todas son altamente exotérmicas), pero su dinámica (su cinética y mecanismos) no podría ser más diferente.
|
|
|
|
|
Los experimentos cuidadosos, llevados a cabo a lo largo de muchos años, son consistentes con el mecanismo más simple: una sola colisión entre las dos moléculas reaccionantes da como resultado un reordenamiento de los enlaces: |
Uno podría tener la tentación de suponer que esto procedería de manera similar, pero los experimentos revelan que el mecanismo de esta reacción es mucho más complejo. La reacción tiene lugar en una sucesión de pasos, algunos de los cuales involucran H atómicos y Br. |
El mecanismo de esta reacción vuelve a ser diferente. Aunque las dos primeras reacciones alcanzan el equilibrio en minutos a una hora más o menos a temperaturas de 300 a 600 K, una mezcla de hidrógeno y cloro no reaccionará en absoluto en la oscuridad, pero si haces brillar una luz sobre la mezcla, se apaga con un estallido ya que la reacción instantánea libera calor y expande el gas explosivamente. |
Lo que es particularmente notable es que estas diferencias llamativas no pueden predecirse de manera confiable a partir de la teoría; solo fueron reveladas por experimentación.
Las tasas de reacciones químicas
Las reacciones químicas varían mucho en la velocidad a la que ocurren. Algunos son esencialmente instantáneos, mientras que otros pueden tardar años en alcanzar el equilibrio. La velocidad de una reacción química puede definirse como el cambio en la concentración de una sustancia dividido por el intervalo de tiempo durante el cual se observa este cambio:
\[\color{red} \text{rate} = \dfrac{\Delta \text{concentration}}{\Delta \text{time}} \label{2-1}\]
Para una reacción de la forma A + B → C, la velocidad se puede expresar en términos del cambio en la concentración de cualquiera de sus componentes:
\[\text{rate} = \dfrac{-\Delta[A]}{\Delta t} \label{Eq1a}\]
\[\text{rate} = \dfrac{-\Delta[B]}{\Delta t} \label{Eq1b}\]
\[\text{rate} = \dfrac{\Delta[C]}{\Delta t} \label{Eq1c}\]
en el que Δ [A] es la diferencia entre la concentración de\(A\) over the time interv al\(\Delta t = t_2 – t_1\):
\[Δ[A] = [A]_2 – [A]_1 \label{2-2}\]
Observe los signos menos en Ecuaciones\(\ref{Eq1a}\) y\(\ref{Eq1a}\); la concentración de un reactivo siempre disminuye con el tiempo, por lo que Δ [A] y Δ [B] son ambos negativos. Dado que las tasas negativas no tienen mucho sentido, las tasas expresadas en términos de concentración de un reactivo siempre van precedidas de un signo menos para que la tasa salga positiva.
Consideremos ahora una reacción en la que los coeficientes son diferentes:
\[A + 3B → 2D \]
Es claro que [B] disminuye tres veces más rápidamente que [A], por lo que para evitar ambigüedades a la hora de expresar la tasa en términos de diferentes componentes, es costumbre dividir cada cambio en la concentración por el coeficiente apropiado:
\[\text{rate} = \dfrac{-\Delta[A]}{\Delta t}= \dfrac{-\Delta[B]}{3 \Delta t} = \dfrac{+\Delta[D]}{2\Delta t}\]
Cada uno de los cocientes anteriores es una expresión legítima de la velocidad de esta reacción en particular; todos dan el mismo número. Cuál se emplea al hacer un cálculo es en gran parte una cuestión de conveniencia.
Para la oxidación del amoníaco
\[4 NH_3 + 3O_2 \rightarrow 2 N_2 + 6 H_2O \nonumber\]
se encontró que la tasa de formación de N 2 fue de 0.27 mol L —1 s —1.
- ¿A qué ritmo se estaba formando el agua?
- ¿A qué ritmo se estaba consumiendo amoníaco?
Solución:
- De la ecuación estequiometría, Δ [H 2 O] = 6/2 Δ [N 2], por lo que la tasa de formación de H 2 O es 3 × (0.27 mol L —1 s —1) = 0.81 mol L —1 s —1.
- Se consumen 4 moles de NH 3 por cada 2 moles de N 2 formados, por lo que la tasa de desaparición del amoníaco es de 2 × (0.27 mol L —1 s —1) = 0.54 mol L —1 s —1.
Comentario: Por la forma en que se formula esta pregunta, sería aceptable expresar este último valor como un número negativo.
Tasas instantáneas
La mayoría de las reacciones se ralentizan a medida que se consumen los reactivos. En consecuencia, las tasas dadas por las expresiones mostradas anteriormente tienden a perder su significado cuando se miden en intervalos de tiempo más largos Δ t. Así, para la reacción cuyo progreso se grafica aquí, la tasa real (medida por la concentración creciente de producto) varía continuamente, siendo mayor en el tiempo cero. La velocidad instantánea de una reacción viene dada por la pendiente de una tangente a la concentración-vs. curva de tiempo. En esta parcela se han identificado tres tasas de este tipo.
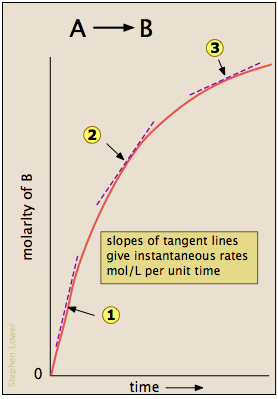
Una velocidad instantánea tomada cerca del inicio de la reacción (t = 0) se conoce como velocidad inicial (etiqueta (1) aquí). Como veremos pronto, las tasas iniciales juegan un papel importante en el estudio de la cinética de reacción. Si has estudiado el cálculo diferencial, sabrás que estas pendientes tangentes son derivadas cuyos valores pueden muy en cada punto de la curva, por lo que estas tasas instantáneas son realmente limitantes las tasas definidas como
\[rate = \lim_{\Delta t \rightarrow 0} \dfrac{-\Delta[A]}{\Delta t}\]
No obstante, si no conoces el cálculo, solo ten en cuenta que cuanto mayor sea el intervalo de tiempo Δ t, menor será la precisión de la tasa instantánea.
Las tasas instantáneas también se conocen como tasas diferenciales.
Leyes de velocidad y orden de reacción
La relación entre la velocidad de una reacción y las concentraciones de reactivos se expresa por su ley de velocidad. Por ejemplo, la velocidad de descomposición en fase gaseosa del pentóxido de dinitrógeno
\[2N_2O_5 → 4NO_2 + O_2\]
se ha encontrado que es directamente proporcional a la concentración de\(N_2O_5\):
\[\text{rate} = k [N_2O_5]\]
Tenga mucho cuidado al confundir las expresiones constantes de equilibrio con las de las leyes de tarifas. La expresión for siempre se\(K_{eq}\) puede escribir inspeccionando la ecuación de reacción, y contiene un término para cada componente (elevado a la potencia apropiada) cuya concentración cambia durante la reacción. Para esta reacción viene dada por
\[ K_{eq} = \dfrac{[NO_2]^2 [O_2]}{[N_2O_5]^2}\]
En contraste, la expresión para la ley de velocidad generalmente no tiene relación necesaria con la ecuación de reacción, y debe determinarse experimentalmente. De manera más general, para una reacción de la forma
\[n_A A + n_B B + ... → products\]
la ley tarifaria será
\[rate = [A]^a[B]^b ... \]
, n B.Dado que la velocidad de una reacción tiene las dimensiones de (concentración/tiempo), las dimensiones de la constante de velocidad k dependerán de los exponentes de los términos de concentración en la ley de velocidad. Para que esto funcione correctamente, si dejamos\(p\) ser la suma de los exponentes de los términos de concentración en la ley de tasas
\[p = a + b + ...\]
entonces k tendrá las dimensiones (concentración 1—p /tiempo).
Orden de reacción
El orden de una ley de tasas es la suma de los exponentes en sus términos de concentración. Para la descomposición de N 2 O 5 con la ley de velocidad k [N 2 O 5], este exponente es 1 (y por lo tanto no se muestra explícitamente); esta reacción es por lo tanto una reacción de primer orden. También podemos decir que la reacción es de “primer orden en N 2 O 5”. Para leyes de tarifas más complicadas, podemos hablar del orden general de reacción y también de las órdenes con respecto a cada componente. Como ejemplo, considere una reacción
\[A + 3B + 2C → \text{products}\]
cuya ley de tasa experimental es
\[rate = k[A] [B]^2\]
Describiríamos esta reacción como
- general de tercer orden,
- de primer orden en A,
- segundo orden en B, y
- orden cero en C.
Orden cero significa que la tasa es independiente de la concentración de un reactivo particular. Sin embargo, debe estar presente suficiente C para permitir que se forme la mezcla de equilibrio.
La velocidad de oxidación de iones bromuro por bromato en una solución acuosa ácida
\[6H^+ + BrO_3^– + 5Br^– → 3 Br_2 + 3 H_2O \nonumber\]
se encuentra que sigue la ley de tarifas
\[rate = k[Br^–][BrO_3^–][H^+]^2\]
¿Qué pasa con la tasa si, en experimentos separados,
- [BrO 3 —] se duplica;
- el pH se incrementa en una unidad;
- la solución se diluye al doble de su volumen, con el pH mantenido constante mediante el uso de un tampón?
Solución
- Dado que la velocidad es de primer orden en bromato, duplicar su concentración duplicará la velocidad de reacción.
- Aumentar el pH en una unidad disminuirá el [H +] en un factor de 10. Dado que la reacción es de segundo orden en [H +], esto disminuirá la tasa en un factor de 100.
- La dilución reduce las concentraciones de Br 2 y BrO 3 — a la mitad de sus valores originales. Hacer esto a cada concentración por sí sola reduciría la tasa en un factor de 2, por lo que reducir ambas concentraciones reducirá la tasa en un factor de 4, a (½) × (½) = ¼ de su valor inicial.
Cómo se observan las órdenes de reacción
Tasa de observación vs. -proporcionalidad de concentración
Para determinar el valor del exponente en un término de ecuación de tasa, necesitamos ver cómo varía la tasa con la concentración de la sustancia. Para una reacción de descomposición de un solo reactivo de la forma
A → productos
en el que la tasa es — d [A]/dt, simplemente trazamos [A] como función del tiempo, dibujamos tangentes a diversos intervalos, y vemos cómo las pendientes de estas tangentes (las tasas instantáneas) dependen de [A].
- Si duplicar la concentración de A duplica la velocidad, entonces la reacción es de primer orden en A.
- Si duplicar la concentración da como resultado un aumento de velocidad de cuatro veces, la reacción es de segundo orden en A.
Utilizar los datos experimentales tabulados para determinar el orden de la reacción
\[2 N_2O_5 → 4 NO_2 + O_2 \nonumber\]
| Tiempo (min) | p (N 2 O 5) | [N 2 O 5] mol L -1 |
Tasa (mol L -1 min -1) |
|---|---|---|---|
| 0 | 301.6 | 0.0152 | |
| 10 | 224.8 | 0.0113 | 3.4 × 10 —4 |
| 20 | 166.7 | 0.0084 | 2.5 |
| 30 | 123.2 | 0.0062 | 1.8 |
| 40 | 92.2 | 0.0046 | 1.3 |
| 69.1 | 69.1 | 0.0035 | 1.0 |
Solución
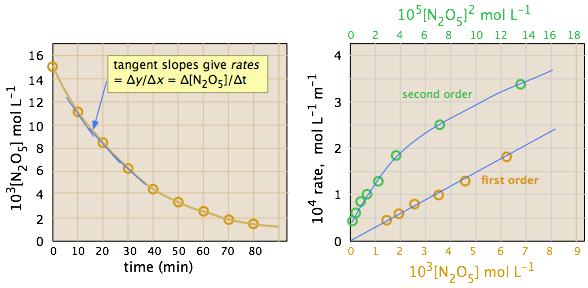
La ley de gas ideal se puede utilizar para convertir las presiones parciales de\(N_2O_5\) a concentraciones molares. Estos son luego trazados (izquierda) para seguir su disminución con el tiempo. Las tasas se calculan a partir de las pendientes de las tangentes (líneas azules) y sus valores se trazan en función de\([N_2O_5]\) y\([N)2O)5]^2\). Es evidente que las tasas son directamente proporcionales a\([N)2O)5]^1\), lo que indica que se trata de una reacción de primer orden.
Método de tasa inicial
Cuando hay más de un reactivo, el método descrito anteriormente rara vez es práctico, ya que las concentraciones de los diferentes reactivos generalmente caerán a diferentes velocidades, dependiendo de la estequiometría. En cambio, medimos solo la velocidad cerca del inicio de la reacción, antes de que las concentraciones hayan tenido tiempo de cambiar significativamente. Luego se repite el experimento con una concentración inicial diferente del reactivo en cuestión, pero manteniendo las concentraciones de cualquier otro igual. Después de encontrar el orden con respecto a un componente, se realiza otra serie de ensayos en los que se encuentra el orden de otro componente.
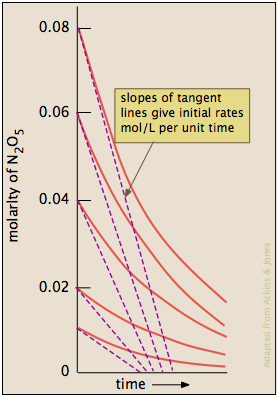

Luego trazamos las cinco tasas iniciales de consumo de\(N_2O_5\) como función de su concentración molar. Como antes, vemos que estas tarifas son directamente proporcionales a\([N_2O_5]\). La pendiente de esta parcela da el valor de la constante de velocidad.
tasa = (5.2 × 10 —3) [N2O5] mol L —1 s —1
Un estudio de la reducción en fase gaseosa del óxido nítrico por hidrógeno
\[2 NO + 2 H_2 → N_2 + 2 H_2O \nonumber\]
arrojó los siguientes datos de velocidad inicial (todas las presiones en torr):
| experimento | P (NO) | P (H 2) |
tasa inicial (torr s-1) |
|---|---|---|---|
|
|
359 | 300 | 1.50 |
|
|
300 | 300 | 1.03 |
|
|
152 | 300 | 0.25 |
|
|
300 | 289 | 1.00 |
|
|
300 | 205 | 0.71 |
|
|
200 | 147 | 0.51 |
Encontrar el orden de la reacción con respecto a cada componente. Al revisar estos datos, tome nota de lo siguiente:
- Las seis corridas aquí registradas se dividen en dos grupos, en los que las presiones iniciales de H 2 y de NO, respectivamente, se mantienen constantes.
- Todos los datos se expresan en presiones, más que en concentraciones. Podemos hacer esto porque los reactivos son gases, cuyas concentraciones son directamente proporcionales a sus presiones parciales cuando T y V se mantienen constantes. Y como sólo nos interesa comparar los ratios de presiones y tasas, las unidades se cancelan y noimporta. Es mucho más fácil ajustar y medir las presiones experimentalmente que las concentraciones.
Solución
Experimentos 2 y 3: La reducción de la presión parcial inicial de NO por un factor de aproximadamente 2 (300/152) da como resultado una reducción de la velocidad inicial por un factor de aproximadamente 4, por lo que la reacción es de segundo orden en óxido nítrico.
Experimentos 4 y 6: Reducir la presión parcial inicial de hidrógeno en un factor de aproximadamente 2 (289/147) provoca una reducción similar en la velocidad inicial, por lo que la reacción es de primer orden en hidrógeno.
La ley de tarifas es así
\[rate = k[NO]^2[H_2]\]
Tratar con múltiples reactivos: el método de aislamiento
No siempre es práctico determinar los órdenes de dos o más reactivos por el método ilustrado en el ejemplo anterior. Afortunadamente, hay otra manera de lograr la misma tarea: podemos usar concentraciones excesivas de todos los reactivos excepto la que deseamos investigar. Por ejemplo, supongamos que la reacción es
\[A + B + C → \text{products}\]
y necesitamos encontrar el orden con respecto a [B] en la ley de tarifas. Si establecemos [B] o en 0.020 M y dejamos [A] o = [C] o = 2.00M, entonces si la reacción va a completarse, el cambio en [A] y [C] también será 0.020 M que es solo el 1 por ciento de sus valores originales. Esto a menudo será menor que el error experimental en la determinación de las tasas, por lo que se puede descuidar. Al “inundar” la mezcla de reacción con uno o más reactivos, estamos aislando efectivamente aquel en el que nos interesa.