
17.3: Colisión y activación- la ley Arrhenius
- Page ID
- 70618

Asegúrese de comprender a fondo las siguientes ideas esenciales que se han presentado anteriormente. Es especialmente imortante que conozcas los significados precisos de todos los términos resaltados en verde en el contexto de este tema.
- Explicar el significado de un mecanismo de reacción y definir paso elemental e intermedio.
- Describir el papel de las colisiones en los mecanismos de reacción y explicar por qué no todas las colisiones conducen a la formación de productos.
- Esbozar diagramas de energía de activación para reacciones simples que son endotérmicas o exotérmicas,
- Explicar en qué se diferencia un complejo activado de un intermedio.
- Defina el catalizador y esboce un diagrama de energía de activación que ilustra cómo funcionan los catalizadores.
- Explicar la significación de los diversos términos que aparecen en la Ley Arrhenius.
- Esbozar una gráfica típica de Arrhenius Law para una reacción hipotética a temperaturas más altas y más bajas.
- Explicar cómo se puede determinar experimentalmente la energía de activación de una reacción.
- Explicar la significación de los diversos términos que aparecen en el factor preexponencial de la ecuación de Arrhenius.
¿Por qué algunas reacciones son mucho más rápidas que otras y por qué las velocidades de reacción son independientes de la tendencia termodinámica de la reacción a tener lugar? Estas son las preguntas centrales que abordamos en esta dependencia. Al hacerlo, abrimos la puerta al importante tema de los mecanismos de reacción: ¿qué sucede a nivel microscópico cuando ocurren reacciones químicas? ¡Podemos agradecer al Prof. Svante Arrhenius por desbloquear esta puerta! Para que las cosas sean lo más simples posible, nos limitaremos a las reacciones que tengan lugar en la fase gaseosa. Los mismos principios se aplicarán a las reacciones en líquidos y sólidos, pero con complicaciones agregadas que discutiremos en una unidad posterior.
Mecanismos de reacción
El mecanismo de una reacción química es la secuencia de eventos reales que ocurren a medida que las moléculas reaccionantes se convierten en productos. Cada uno de estos eventos constituye un paso elemental que puede representarse como una unión de partículas discretas (“collison”) o como la ruptura de una molécula (“disociación”) en unidades más simples. La entidad molecular que emerge de cada etapa puede ser un producto final de la reacción, o podría ser un intermedio, una especie que se crea en un paso elemental y se destruye en una etapa posterior, y por lo tanto no aparece en la ecuación de reacción neta.
Un mecanismo de reacción debe entenderse en última instancia como una descripción “soplada a golpe” de los eventos a nivel molecular cuya secuencia conduce de reactivos a productos. Estos pasos elementales (también llamados reacciones elementales) son casi siempre muy simples que involucran una, dos o [raramente] tres especies químicas que se clasifican, respectivamente, como
| unimolecular | A → | con mucho el más común |
| bimolecular | A + B → | |
| termolecular | A + B + C → | muy raro |
Teoría de colisión del cambio químico: Las moléculas deben colisionar antes de que puedan reaccionar
Esta regla fundamental debe guiar cualquier análisis de un mecanismo ordinario de reacción química.

Esto explica por qué los procesos termoleculares son tan poco comunes. La teoría cinética de los gases nos dice que por cada 1000 colisiones binarias, solo habrá un evento en el que tres moléculas se juntan simultáneamente. Las colisiones de cuatro vías son tan improbables que este proceso nunca se ha demostrado en una reacción elemental. Considera un simple paso bimolecular
\[\ce{A + B -> products} \nonumber\]
Claramente, si dos moléculas A y B van a reaccionar, deben acercarse lo suficientemente cerca como para perturbar algunos de sus vínculos existentes y permitir la creación de cualesquiera nuevas que sean necesarias en los productos. Llamamos a tal encuentro una colisión.
La frecuencia de colisiones entre A y B en un gas será proporcional a la concentración de cada uno; si doblamos [A], la frecuencia de colisiones A-B se duplicará, y duplicar [B] tendrá el mismo efecto. Entonces, si todas las colisiones conducen a productos, entonces la tasa de un proceso bimolecular será de primer orden en A y B, o de segundo orden general:
\[\text{rate} = k[\ce{A}][\ce{B}] \nonumber\]
Sin embargo, no todas las colisiones son iguales. En un gas a temperatura ambiente y presión atmosférica normal, habrá alrededor de 10 33 colisiones en cada centímetro cúbico cada segundo. Si cada colisión entre dos moléculas reaccionantes produjera productos, todas las reacciones estarían completas en una fracción de segundo.
Cuando dos bolas de billar chocan, simplemente rebotan una de la otra. Este es también el resultado más probable si la reacción entre A y B requiere una alteración o reordenamiento significativo de los enlaces entre sus átomos. Para iniciar efectivamente una reacción, las colisiones deben ser suficientemente energéticas (energía cinética) para provocar esta interrupción del enlace. Más sobre esto más adelante.
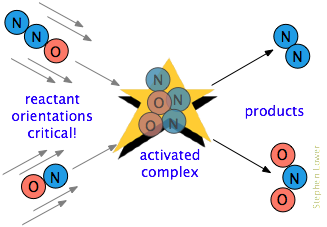
Y a menudo hay un requisito adicional. En muchas reacciones, especialmente aquellas que involucran moléculas más complejas, las especies reaccionantes deben orientarse de una manera apropiada para el proceso particular. Por ejemplo, en la reacción en fase gaseosa del óxido de dinitrógeno con óxido nítrico, el extremo de oxígeno del N 2 O debe golpear el extremo nitrógeno del NO; invertir la orientación de cualquiera de las moléculas impide la reacción.
Debido a la amplia aleatorización de movimientos moleculares en un gas o líquido, siempre hay suficientes moléculas orientadas correctamente para que algunas de las moléculas reaccionen. Pero claro, cuanto más crítico sea este requisito orientacional, menos colisiones serán efectivas.
Anatomía de una colisión
Las colisiones energéticas entre moléculas hacen que los enlaces interatómicos se estiren y se doblen más, debilitándolos temporalmente para que se vuelvan más susceptibles a la escisión. La distorsión de los enlaces puede exponer sus nubes de electrones asociadas a interacciones con otros reactivos que podrían conducir a la formación de nuevos enlaces. Los enlaces químicos tienen algunas de las propiedades de los resortes mecánicos, cuya energía potencial depende de la medida en que se estiren o compriman. Cada enlace átomo a átomo puede describirse mediante un diagrama de energía potencial que muestra cómo cambia su energía con su longitud. Cuando el enlace absorbe energía (ya sea del calentamiento o a través de una colisión), se eleva a un estado vibracional cuantificado más alto (indicado por las líneas horizontales) que debilita el enlace a medida que su longitud oscila entre los límites extendidos correspondientes a la curva.
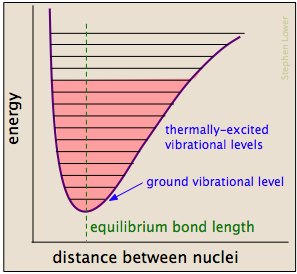
Una colisión particular normalmente excitará una serie de enlaces de esta manera. En aproximadamente 10 —13 segundos esta excitación se distribuye entre los otros enlaces de la molécula de formas bastante complejas e impredecibles que pueden concentrar la energía agregada en un punto particularmente vulnerable. El enlace afectado puede estirarse y doblarse más lejos, haciéndolo más susceptible a la escisión. Incluso si el enlace no se rompe por estiramiento puro, puede distorsionarse o retorcerse para exponer las nubes de electrones cercanas a interacciones con otros reactivos que podrían alentar una reacción.
Consideremos, por ejemplo, la isomerización del ciclopropano a propeno que tiene lugar a temperaturas bastante altas en la fase gaseosa.

Podemos imaginar la secuencia de colisión-a-producto de la siguiente manera [muy simplificada]:
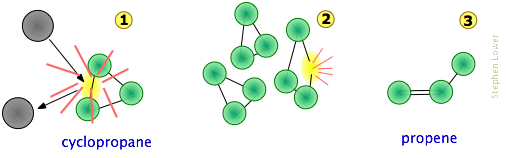
Tenga en cuenta que
- Para que las cosas sean simples, aquí no mostramos los átomos de hidrógeno. Esto es razonable porque los enlaces C-C son más débiles que los enlaces C—H y, por lo tanto, es menos probable que se vean afectados.
- La colisión a
voluntad suele ser con otra molécula de ciclopropano, pero debido a que ninguna parte de la molécula colisionante se incorpora al producto, en principio puede ser un gas noble o alguna otra especie no reaccionante;
- Aunque los enlaces C-C en el ciclopropano son todos identiciales, la localización instantánea de la energía colisional puede distorsionar la molécula de varias maneras (
Los procesos unimoleculares también comienzan con una colisión
La isomerización de ciclopropano descrita anteriormente es típica de muchas reacciones de descomposición que se encuentran que siguen cinéticas de primer orden, lo que implica que el proceso es unimolecular. Hasta alrededor de 1921, los químicos no entendieron el papel de las colisiones en procesos unimoleculares. Resulta que los mecanismos de tales reacciones son realmente bastante complicados, y que a presiones muy bajas sí siguen una cinética de segundo orden. Tales reacciones se describen más adecuadamente como pseudounimoleculares.
Energía de activación
Temperaturas más altas, reacciones más rápidas
Las reacciones químicas asociadas con la mayor parte del deterioro de los alimentos son catalizadas por enzimas producidas por las bacterias que median estos procesos. Es de conocimiento común que las reacciones químicas ocurren más rápidamente a temperaturas más altas. Todo el mundo sabe que la leche se vuelve agria mucho más rápidamente si se almacena a temperatura ambiente en lugar de en un refrigerador, la mantequilla se vuelve rancia más rápido en verano que en invierno, y los huevos hierven más rápidamente al nivel del mar que en las montañas. Por la misma razón, los animales de sangre fría como reptiles e insectos tienden a ser notablemente más letárgicos en los días fríos.
No es difícil entender por qué debería ser esto. La energía térmica relaciona la dirección con el movimiento a nivel molecular. A medida que aumenta la temperatura, las moléculas se mueven más rápido y chocan más vigorosamente, aumentando en gran medida la probabilidad de escisiones y reordenamientos de enlaces como se describió anteriormente.
Diagramas energéticos de activación
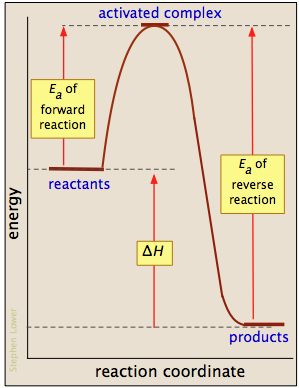
La mayoría de las reacciones que involucran moléculas neutras no pueden tener lugar en absoluto hasta que hayan adquirido la energía necesaria para estirar, doblar o distorsionar uno o más enlaces. Esta energía crítica se conoce como la energía de activación de la reacción. Los diagramas de energía de activación del tipo que se muestran a continuación representan la entrada de energía total a un sistema de reacción a medida que avanza de los reactivos a los productos.
Al examinar dichos diagramas, tome nota especial de lo siguiente:
- La "coordenada de reacción" trazada a lo largo de la abscisa representa los cambios en las coordenadas atómicas a medida que el sistema avanza de reactivos a productos. En las reacciones elementales muy simples podría corresponder al estiramiento o torsión de un enlace particular, y mostrarse a una escala. En general, sin embargo, la coordenada de reacción es un concepto bastante abstracto que no puede vincularse a ninguna cantidad medible y escalable.
- El complejo activado (también conocido como estado de transición) representa la estructura del sistema tal como existe en el pico de la curva de energía de activación. No se corresponde con una estructura intermedia identificable (que más adecuadamente se consideraría el producto de un proceso elemental separado), sino a cualquier configuración de átomos que exista durante la colisión, que dura solo alrededor de 0.1 picosegundos.
- Los diagramas de energía de activación siempre incorporan la energía (Δ U o Δ H) de la reacción neta, pero es importante entender que estas últimas cantidades dependen únicamente de la termodinámica del proceso que siempre son independientes de la vía de reacción. Esto significa que la misma reacción puede exhibir diferentes energías de activación si puede seguir vías alternativas.
- Con algunas excepciones para procesos muy simples, los diagramas de energía de activación son en gran parte constructos conceptuales basados en nuestro modelo de colisión estándar para reacciones químicas. Sería imprudente leer demasiado en ellos.
Galería de parcelas de energía de activación
Los diagramas de energía de activación pueden describir reacciones exotérmicas y endotérmicas:
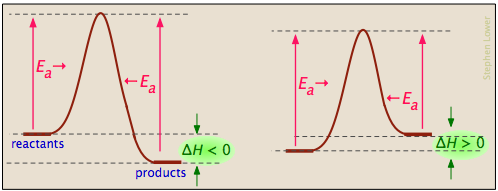
... y las energías de activación de la reacción directa pueden ser grandes, pequeñas o cero (independientemente, por supuesto, del valor de Δ H):
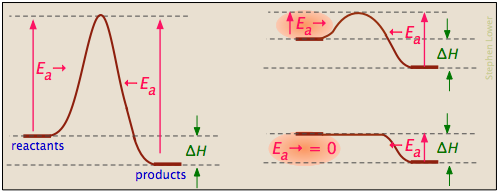
Los procesos con energía de activación cero suelen implicar la combinación de iones con carga opuesta o el emparejamiento de electrones en radicales libres, como en la dimerización del óxido nítrico (que es una molécula de electrones impar).
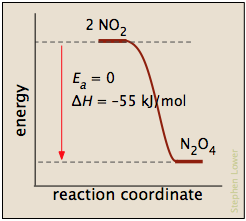
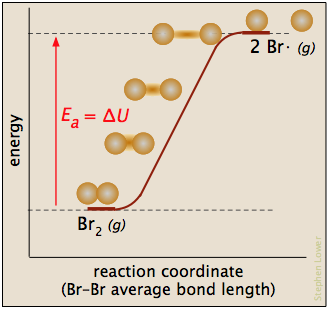
En esta gráfica para la disociación de bromo, la E a es solo la entalpía de atomización
Br 2 (g) → 2 Br· (g)
y la coordenada de reacción corresponde aproximadamente al estiramiento del enlace vibracionalmente excitado. El “complejo activado”, si se considera que existe, es apenas el último, más largo “tramo”. La reacción inversa, siendo la recombinación de dos radicales, ocurre inmediatamente al contacto.
¿De dónde viene la energía de activación?
En la mayoría de los casos, la energía de activación es suministrada por energía térmica, ya sea a través de colisiones intermoleculares o (en el caso de dissocación térmica) por excitación térmica de una vibración de estiramiento de enlace a un nivel cuántico suficientemente alto.
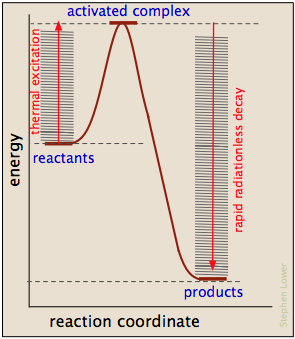
A medida que se forman los productos, la energía de activación se devuelve en forma de energía vibratoria que se degrada rápidamente a calor. Vale la pena señalar, sin embargo, que a veces son aplicables otras fuentes de energía de activación:
- La absorción de luz por una molécula (fotoexcitación) puede ser una muy limpia y eficiente, pero no siempre funciona. No es suficiente que la longitud de onda de la luz corresponda a la energía de activación; también debe caer dentro del espectro de absorción de la molécula, y (en una molécula compleja) suficiente de ella debe terminar en la parte derecha de la molécula, como en un enlace particular.
- Activación electroquímica. Las moléculas capaces de perder o ganar electrones en la superficie de un electrodo pueden sufrir activación a partir de un potencial extra (conocido como la sobretensión) entre el electrodo y la solución. La superficie del electrodo a menudo juega un papel activo, por lo que el proceso también se conoce como electrocatálisis.
Los catalizadores pueden reducir la energía de activación
Un catalizador suele definirse como una sustancia que acelera una reacción sin ser consumida por él. Más específicamente, un catalizador proporciona una vía alternativa de energía de activación más baja entre los reactivos y los productos. Como tales, son de vital importancia para la tecnología química; aproximadamente el 95% de los procesos químicos industriales involucran catalizadores de diversos tipos. Además, la mayoría de los procesos bioquímicos que ocurren en los organismos vivos están mediados por enzimas, que son catalizadores hechos de proteínas.
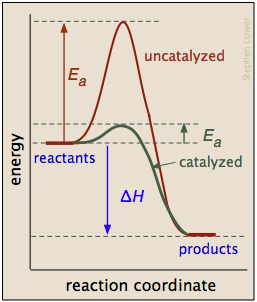
Es importante entender que un catalizador afecta solo la cinética de una reacción; no altera la tendencia termodinámica para que se produzca la reacción. Así, existe un único valor de Δ H para las dos vías representadas en la gráfica de la derecha
Temperatura y energía cinética
Una revisión de los principios de las velocidades moleculares del gas y la distribución de Boltzmann se puede encontrar en la página “KMT-Classic”.
En la gran mayoría de los casos, dependemos de la actvación térmica, por lo que el factor principal que debemos considerar es qué fracción de las moléculas posee suficiente energía cinética para reaccionar a una temperatura dada. Según la teoría molecular cinética, una opulación de moléculas a una temperatura dada se distribuye sobre una variedad de energías cinéticas descritas por la ley de distribución de Maxwell-Boltzman.
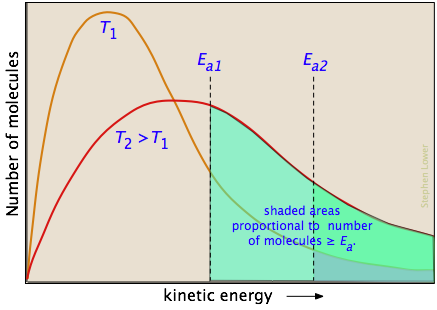
Las dos gráficas de distribución que se muestran aquí son para una temperatura T 1 más baja y una temperatura T 2 más alta. El área bajo cada curva representa el número total de moléculas cuyas energías caen dentro de un rango particular. Las regiones sombreadas indican el número de moléculas que son suficientemente energéticas para cumplir con los requisitos dictados por los dos valores de E a que se muestran.
Es claro a partir de estas gráficas que la fracción de moléculas cuya energía cinética excede la energía de activación aumenta bastante rápidamente a medida que se eleva la temperatura. Esta es la razón por la que prácticamente todas las reacciones químicas (y todas las reacciones elementales) son más rápidas a temperaturas más altas.
2 La ley Arrhenius
Para 1890 era de conocimiento común que las temperaturas más altas aceleran las reacciones, a menudo duplicando la tasa para un aumento de 10 grados, pero las razones de esto no estaban claras. Finalmente, en 1899, el químico sueco Svante Arrhenius (1859-1927) combinó los conceptos de energía de activación y la ley de disribución de Boltzmann en una de las relaciones más importantes en la química física:

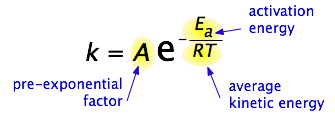
Tómate un momento para enfocarte en el significado de esta ecuación, descuidando por el momento el factor A.
Primero, señalar que esta es otra forma de la ley de decaimiento exponencial que discutimos en la sección anterior de esta serie. Lo que aquí es “decayendo” no es la concentración de un reactivo en función del tiempo, sino la magnitud de la constante de velocidad en función del exponente —E a/RT. ¿Y cuál es el significado de esta cantidad? Si recuerdas que RT es la energía cinética promedio, será evidente que el exponente es solo la relación de la energía de activación E a la energía cinética promedio. Cuanto mayor sea esta relación, menor será la tasa (de ahí el signo negativo). Esto significa que la alta temperatura y la baja energía de activación favorecen mayores constantes de velocidad y, por lo tanto, aceleran la reacción. Y debido a que estos términos ocurren en un exponente, sus efectos sobre la tasa son bastante sustanciales.
Las dos gráficas siguientes muestran los efectos de la energía de activación (denotada aquí por E ‡) sobre la constante de velocidad. Incluso una modesta energía de activación de 50 kJ/mol reduce la tasa en un factor de 10 8.

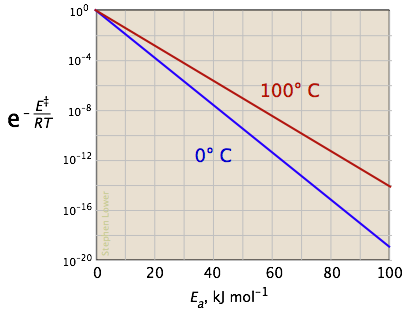
La escala logarítmica en la gráfica de la derecha conduce a bonitas líneas rectas, como se describe bajo el siguiente encabezamiento a continuación.
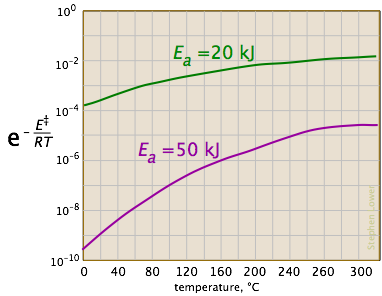
Al observar el papel de la temperatura, vemos un efecto similar. (Si el eje x estuviera en “kilogrados” las pendientes serían más comparables en magnitud con las de la parcela de kilojulios de arriba a la derecha).
Determinar la energía de activación
La ecuación de Arrhenius
\[k=A \mathrm{e}^{-E_{a} / R T}\]
se puede escribir en una forma no exponencial que a menudo es más conveniente de usar e interpretar gráficamente. Tomando los logaritmos de ambos lados y separando los términos exponenciales y preexponenciales rendimientos
\[\ln k=\ln \left(A \mathrm{e}^{-E_{\mathrm{a}} / R T}\right)=\ln A+\ln \left(\mathrm{e}^{-E_{\mathrm{a}} / R T}\right)\]
\[\ln k=\ln A-\dfrac{E_{a}}{R T}\]
que es la ecuación de una línea recta cuya pendiente es\(–E_a /R\). Esto permite una forma sencilla de determinar la energía de activación a partir de valores\(k\) observados a diferentes temperaturas; solo trazamos\(\ln k\) como una función de\(1/T\).
Así, para la isomerización de ciclopropano a propeno
se obtuvieron los siguientes datos (valores calculados sombreados en rosa):

| T, °C | 477 | 523 | 577 | 623 |
|---|---|---|---|---|
| 1/ T, K —1 × 10 3 | 1.33 | 1.25 | 1.18 | 1.11 |
| k, s —1 | 0.00018 | 0.0027 | 0.030 | 0.26 |
| ln k | —8.62 | —5.92 | —3.51 | —1.35 |
A partir de la pendiente calculada, tenemos
— (E a/R) = —3.27 × 10 4 K
E a =— (8.314 J mol —1 K —1) (—3.27 × 10 4 K) = 273 kJ mol —1
Comentario: Esta energía de activación es bastante alta, lo cual no es sorprendente porque se debe romper un enlace carbono-carbono para abrir el anillo de ciclopropano. (Las energías de enlace C-C son típicamente de alrededor de 350 kJ/mol.) Es por ello que la reacción debe llevarse a cabo a alta temperatura.
No siempre necesitas una parcela
(... ¡si estás dispuesto a vivir un poco peligrosamente!) Dado que la gráfica ln k -vs.-1/ T produce una línea recta, a menudo es conveniente estimar la energía de activación a partir de experimentos a solo dos temperaturas. Para ver cómo se hace esto, considera que
\[\ln k_{2}-\ln k_{1}=\left(\ln A-\frac{E_{a}}{R T_{2}}\right)-\left(\ln A-\frac{E_{a}}{R T_{1}}\right)=\frac{E_{a}}{R}\left(\frac{1}{T_{1}}-\frac{1}{T_{2}}\right) \nonumber\]
(... en la que hemos hecho desaparecer el término ln- A restando las expresiones para los dos términos ln- k.) Resolviendo la expresión a la derecha para los rendimientos de energía de activación
\[E_{a}=\dfrac{R \ln \dfrac{k_{2}}{k_{1}}}{\dfrac{1}{T_{1}}-\dfrac{1}{T_{2}}} \nonumber\]
Una regla general ampliamente utilizada para la dependencia de la temperatura de una velocidad de reacción es que un aumento de diez C° en la temperatura aproximadamente duplica la velocidad. (Esto obviamente no es generalmente cierto, especialmente cuando se debe romper un enlace covalente fuerte). Pero para una reacción que sí muestre este comportamiento, ¿cuál sería la energía de activación?
Solución
Centraremos nuestro intervalo de diez grados en 300 K. Sustituyendo en los rendimientos de expresión anteriores
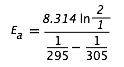 = (8.314) (0.693)/(.00339 - 0.00328)
= (8.314) (0.693)/(.00339 - 0.00328)
= (5.76 J mol —1 K —1)/(0.00011 K —1) = 52400 J mol —1 = 52.4 kJ mol —1
Se tarda unos 3.0 minutos en cocinar un huevo duro en Los Ángeles, pero a la mayor altitud de Denver, donde el agua hierve a 92°C, el tiempo de cocción es de 4.5 minutos. Utilice esta información para estimar la energía de activación para la coagulación de la proteína de albúmina de huevo.
Solución:
La relación de las constantes de velocidad en las elevaciones de LA y Denver es 4.5/3.0 = 1.5, y las temperaturas respectivas son 373K y 365K. Con los subíndices 2 y 1 referidos a LA y Denver respectivamente, tenemos
E a = (8.314) (ln 1.5)/(1/365 — 1/273) = (8.314) (.405)/(0.00274 — 0.00366)
= (3.37 J mol —1 K —1)/(0.000923 K —1) = 3650 J mol —1 = 3.65 kJ mol —1
Comentario: Este valor bastante bajo parece razonable porque la desnaturalización de proteínas implica la alteración de enlaces de hidrógeno relativamente débiles; no se rompen enlaces covalentes
Grillos y palomitas
- Muchos procesos biológicos exhiben una dependencia de la temperatura que sigue la ley de Arrhenius y, por lo tanto, pueden caracterizarse por una energía de activación. Mira esta interesante página de Dartmouth U. que analiza la cinética de los chirps de cricket.
El factor preexponencial
Ahora es el momento de enfocarse en el término preexponencial A en la ecuación de Arrhenius. La hemos estado descuidando porque no está directamente involucrada en relacionar la temperatura y la energía de activación, que es el principal uso práctico de la ecuación. Pero como A multiplica el término exponencial, su valor contribuye claramente al valor de la constante de tasa y por lo tanto de la tasa.
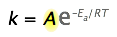
y de la temperatura.
Si esta fracción fuera unidad, la ley Arrhenius reduciría a
\[k = A\]
En otras palabras, A es la fracción de moléculas que reaccionarían si o bien la energía de activación fuera cero, o si la energía cinética de todas las moléculas excediera a E a —ciertamente, un escenario poco común.
Se trata de colisiones
Entonces, ¿qué limitaría la constante de velocidad si no hubiera requisitos de energía de activación? El factor más obvio sería la velocidad a la que las moléculas reaccionantes entran en contacto. Esto se puede calcular a partir de la teoría molecular cinética y se conoce como el factor de frecuencia o colisión Z. En algunas reacciones, la orientación relativa de las moléculas en el punto de colisión es importante, por lo que también podemos definir un factor geométrico o estérico (comúnmente denotado por ρ (rho minúsculas griegas).
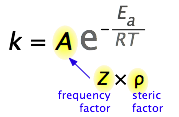
En general, podemos expresar A como producto de estos dos factores:
\[A = Zρ\]
Los valores de generalmente\(ρ\) son muy difíciles de evaluar; en algún momento se estiman comparando la constante de velocidad observada con aquella en la que A se supone que es la misma que Z.
La dirección marca la diferencia
Cuanto más complicadas sean las estructuras de los reactivos, más probable es que el valor de la constante de velocidad dependa de las trayectorias en las que los reactivos se acercan entre sí. Mostramos un ejemplo de esto cerca de la parte superior de la página, pero para otro, consideremos la adición de un haluro de hidrógeno como HCl al doble enlace de un alqueno, convirtiéndolo en cloroalcano. Este tipo de reacción de adición electrófila es bien conocida por todos los estudiantes de química orgánica.
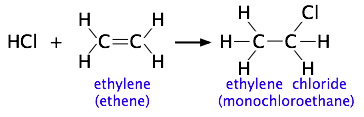
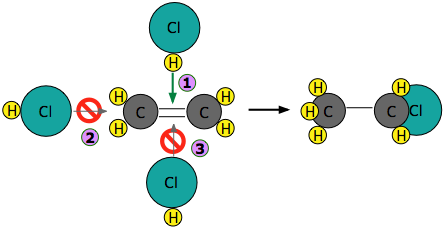
Los experimentos han demostrado que la reacción solo tiene lugar cuando la molécula de HCl se acerca al alqueno con su extremo de hidrógeno, y en una dirección que es aproximadamente perpendicular al doble enlace, como se muestra a continuación.
continuación.
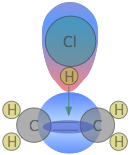
La razón de esto se hace evidente cuando recordamos que el HCl es altamente polar debido a la alta electronegatividad del cloro, por lo que el extremo de hidrógeno de la molécula es ligeramente positivo. El doble enlace del eteno consiste en dos nubes de carga negativa correspondientes a los orbitales moleculares σ (sigma) y π (pi). Este último, que se extiende por encima y por debajo del plano de la molécula C 2 H 4, interactúa y atrae a la molécula de HCl.
Si, en cambio, el HCl se acerca con su extremo de cloro conduciendo como en , la repulsión electrostática entre las cargas similares hace que las dos moléculas reboten una de la otra antes de que pueda tener lugar cualquier reacción. Lo mismo sucede en
, la repulsión electrostática entre las cargas similares hace que las dos moléculas reboten una de la otra antes de que pueda tener lugar cualquier reacción. Lo mismo sucede en ; la diferencia de electronegatividad entre carbono e hidrógeno es demasiado pequeña para hacer que el enlace C—H sea suficientemente polar para atraer el átomo de cloro entrante.
; la diferencia de electronegatividad entre carbono e hidrógeno es demasiado pequeña para hacer que el enlace C—H sea suficientemente polar para atraer el átomo de cloro entrante.
La lección que debes tomar de este ejemplo es que una vez que comienzas a combinar una variedad de principios químicos, gradualmente desarrollas lo que podría llamarse “intuición química” que puedes aplicar a una amplia variedad de problemas. Esto es mucho más importante que memorizar ejemplos específicos. Ahora que ya sabes lo que se necesita para comenzar una reacción, estás listo para la siguiente lección que describe sus mecanismos reales.