
17.6: Catalizadores y Catálisis
- Page ID
- 70611

Asegúrese de comprender a fondo las siguientes ideas esenciales que se han presentado anteriormente. Es especialmente imortante que conozcas los significados precisos de todos los términos verde-resaltados en el contexto de este tema.
- ¿Qué son los catalizadores y cómo funcionan en términos alterando los parámetros de una reacción?
- Describir las similitudes y diferencias entre las tres clases principales de catalizadores.
- Definir fisisorción y quimisorción, y explicar el papel de esta última en el inicio de un evento catalítico.
- ¿Cuál es el significado y significado del Principio Sabatier?
- ¿Cuáles son las principales diferencias entre los mecanismos Langmuir-Hinshelwood y Eley-Rideal de catálisis heterogénea?
- Describir el papel del complejo enzima-sustrato.
- ¿Cómo podrían contribuir a su efecto catalítico las propiedades particulares de los aminoácidos que rodean el sitio activo de una enzima?
- Describir los modelos de acción enzimática de bloqueo y llave y ajuste inducido.
- Explicar la función y significancia de los sitios alostéricos en una enzima.
¡Casi parece magia! Una mezcla de H 2 y O 2 gaseosos puede coexistir indefinidamente sin ninguna reacción detectable, pero si se calienta un alambre de platino y luego se sumerge en la mezcla gaseosa, la reacción avanza suavemente a medida que el calor liberado por la reacción hace que el alambre brille al rojo vivo. Los catalizadores juegan un papel esencial en nuestra economía industrial moderna, en nuestra gestión del medio ambiente y en todos los procesos biológicos. Esta lección te dará un vistazo al maravilloso mundo de los catalizadores, ayudándote a entender qué son y cómo funcionan.
¿Qué son los Catalizadores?
Los catalizadores no tienen efecto sobre la constante de equilibrio y, por lo tanto, sobre la composición de equilibrio. Los catalizadores son sustancias que aceleran una reacción pero que no son consumidas por ella y no aparecen en la ecuación de reacción neta. También —y esto es muy importante— los catalizadores afectan las tasas de avance y retroceso por igual; esto significa que los catalizadores no tienen ningún efecto sobre la constante de equilibrio y, por lo tanto, sobre la composición del estado de equilibrio. Así, se puede usar un catalizador (en este caso, ácido sulfúrico) para acelerar una reacción reversible como la formación de éster o su inversa, hidrólisis de éster:
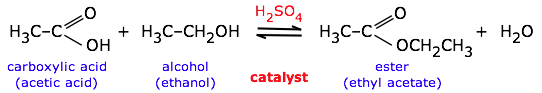
El catalizador no tiene ningún efecto sobre la constante de equilibrio o la dirección de la reacción. La dirección se puede controlar añadiendo o eliminando agua (principio Le Chatelier).
Los catalizadores funcionan permitiendo que la reacción tenga lugar a través de un mecanismo alternativo que requiere una energía de activación más pequeña. Este cambio es provocado por una interacción específica entre el catalizador y los componentes de la reacción. Recordarás que la constante de velocidad de una reacción es una función exponencial de la energía de activación, por lo que incluso una modesta reducción de\(E_a\) puede producir un incremento impresionante en la tasa.
Los catalizadores proporcionan vías de reacción alternativas
Los catalizadores se dividen convencionalmente en dos categorías: homogéneos y heterogéneos. Las enzimas, catalizadores biológicos naturales, a menudo se incluyen en el primer grupo, pero debido a que comparten algunas propiedades de ambos pero exhiben algunas propiedades propias muy especiales, las trataremos aquí como una tercera categoría.
Algunos ejemplos comunes de catálisis
Cómo quemar un Sugar Cube
Cuando se calienta solo, un cubo de azúcar (sacarosa) se funde a 185°C pero no se quema. Pero si el cubo se frota en cenizas de cigarrillo, se quema antes de fundirse debido a la acción catalítica de los compuestos de metales traza en las cenizas.
Platino como Catalizador de Oxidación
La superficie del platino metálico es un catalizador eficiente para la oxidación de muchos vapores de combustible. Esta propiedad se explota en estufas de camping sin llama (izquierda). La imagen de la derecha muestra un brillante alambre de platino calentado por la combustión lenta del amoníaco en su superficie. Sin embargo, si sumerges un cable Pt calentado en amoníaco líquido, obtienes una explosión en miniatura: vea el video a continuación.
Esta es la oxidación del amoníaco por oxígeno catalizada por alambre de platino calentado. El oxígeno se burbujea a través de la solución de amoníaco, en la que se mezcla con el gas amoníaco presente La reacción hace que el alambre de platino brille, y el alambre caliente enciende una mezcla de amoníaco y oxígeno.
Descomposición del peróxido de hidrógeno
El peróxido de hidrógeno es termodinámicamente inestable según la reacción
\[\ce{2 H2O2 → 2 H2O + O2 } \quad \quad ΔG^o = –210\, kJ\, mol^{–1}\]
En ausencia de contaminantes esta reacción es muy lenta, pero una variedad de sustancias, que van desde yodo, óxidos metálicos, trazas de metales, aceleran enormemente la reacción, en algunos casos casi explosivamente debido a la rápida liberación de calor. El catalizador más efectivo de todos es la enzima catalasa, presente en sangre y fluidos intracelulares; agregar una gota de sangre a una solución de peróxido de hidrógeno al 30% induce una reacción vigorosa.
La rápida liberación de O 2 puede resultar en un espectacular baño de burbujas si se agrega algo de jabón. [¡Esta misma reacción se ha utilizado para alimentar un auto de carreras!
El yoduro de potasio cataliza eficientemente la deposición de H 2 O 2. Este breve video muestra lo que sucede cuando se combinan algún jabón coloreado, H 2 O 2, y KI. ¡Pero “no intentes esto en [tu] casa”!
Cada tipo de catalizador facilita una vía diferente con su propia energía de activación. Debido a que la velocidad es una función exponencial de E a (ecuación de Arrhenius), incluso diferencias relativamente pequeñas en E a pueden tener efectos dramáticos en las velocidades de reacción. Tenga en cuenta especialmente los valores para la catalasa; ¡el químico sigue siendo un aficionado de rango comparado con lo que la naturaleza puede lograr a través de la selección natural!
| catalizador |
Ea |
tasa relativa |
|---|---|---|
| sin catalizador | 75 | 1 |
| ión yoduro | 56 | 2140 |
| Platino coloidal | 50 | 24,000 |
| catalasa (enzima) | 21 | 2,900,000,000 |
Cómo se expresa la actividad catalítica
Los cambios en la constante de velocidad o de la energía de activación son formas obvias de medir la eficacia de un catalizador. Pero han entrado en uso otros dos términos que tienen especial relevancia en aplicaciones industriales.
Número de facturación
El número de recambio (TON) es un número promedio de ciclos que un catalizador puede sufrir antes de que su rendimiento se deteriore (ver más adelante). Las toneladas reportadas para catalizadores industriales comunes abarcan un rango muy amplio de quizás 10 a más de 10 5, lo que se acerca a los límites del transporte de difusión.
Frecuencia de rotación
Este término, que originalmente se aplicó a las reacciones catalizadas por enzimas, ha entrado en uso más general. Es simplemente el número de veces que la reacción catalizada global tiene lugar por catalizador (o por sitio activo en una enzima o catalizador heterogéneo) por unidad de tiempo: se define como
El número de sitios activos S en un catalizador heterogéneo suele ser difícil de estimar, por lo que a menudo es reemplazado por el área total del catalizador expuesto, que generalmente es medible experimentalmente. Los TOF para reacciones heterogéneas generalmente caen entre 10 —2 a 10 2 s —1.
Catálisis Homogénea
Como su nombre lo indica, los catalizadores homogéneos están presentes en la misma fase (solución gaseosa o líquida) que los reactivos. Los catalizadores homogéneos generalmente entran directamente en la reacción química (formando un nuevo compuesto o complejo con un reactivo), pero se liberan en su forma inicial después de que se completa la reacción, de manera que no aparecen en la ecuación de reacción neta.
Isomerización cis-trans catalizada por yodo
A menos que estés tomando un curso de química orgánica en el que tu instructor indique lo contrario, no intentes memorizar estos mecanismos. Se presentan aquí con el propósito de convencerte de que la catálisis no es magia negra, y para familiarizarte con algunas de las características de los mecanismos catalizados. Debería ser suficiente que usted se limite a convencerse de que los pasos individuales tienen sentido químico.
Recordará que el isomerismo cis-trans es posible cuando los átomos conectados a cada uno de los dos carbonos doblemente unidos pueden estar en el mismo (cis) o lados opuestos (trans) del enlace. Esto refleja el hecho de que la rotación alrededor de un doble enlace no es posible.
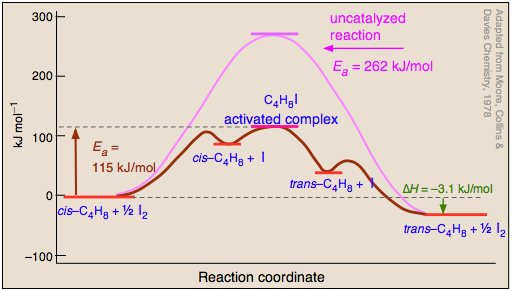
La conversión de un alqueno entre sus formas cis y trans solo puede ocurrir si el doble enlace se rompe temporalmente, liberando así los dos extremos para que roten. Los procesos que escinden enlaces covalentes tienen altas energías de activación, por lo que las reacciones de isomerización cis-trans tienden a ser lentas incluso a altas temperaturas. El yodo es uno de varios catalizadores que aceleran enormemente este proceso, por lo que la isomerización del buteno sirve como un buen ejemplo introductorio de catálisis homogénea.
Se cree que el mecanismo de la reacción catalizada por yodo implica el ataque de átomos de yodo (formados por el equilibrio de disociación en uno de los carbonos en el Paso
:
Durante su breve existencia, el complejo activado por rédica libre puede sufrir rotación alrededor del enlace C-C, de manera que cuando se descompone liberando el yodo (), una porción del buteno reconstituido estará en su forma trans. Finalmente, el átomo de yodo se recombina en diyodo. Dado que procesa
y
cancela, el yodo no aparece en la ecuación de reacción neta, requisito para un verdadero catalizador.
Catálisis ácido-base
Muchas reacciones son catalizadas por la presencia de un ácido o una base; en muchos casos, tanto ácidos como bases catalizarán la misma reacción. Como cabría esperar, el mecanismo implica la adición o eliminación de un protón, cambiando el reactivo a una forma más lábil cinéticamente. Un ejemplo sencillo es la adición de yodo a la propanona
I 2 + (CH 3) 2 —C=O → (CH 2 I) (CH 3) —C=O
El mecanismo para el proceso catalizado por ácido implica varios pasos. El papel del ácido es proporcionar un protón que se une al oxígeno carbonilo, formando un ion oxonio inestable. Este último se reorganiza rápidamente en un enol
(es decir, un carbono conectado tanto a un doble enlace (eno) como a un grupo hidroxilo (ol)). Esto completa la parte catalítica del proceso, que es básicamente una reacción ácido-base (transferencia de protones) en la que el papel del protón es extraer un electrón del oxígeno de la cetona.
En la segunda etapa, el enol reacciona con el yodo. Las flechas curvas indican cambios en las ubicaciones de los electrones. En abajo, un electrón es retirado del orbital π del doble enlace por uno de los átomos de la molécula I 2. Esto induce un desplazamiento de electrones en este último, provocando que la mitad de esta molécula sea expulsada como un ión yoduro. La otra mitad del yodo es ahora un ion yodonio I + que desplaza un protón de uno de los grupos metilo. El ion carbonio resultante expulsa
entonces el protón -OH para producir el producto neutro final.
Quizás la reacción catalizada por ácido más conocida es la hidrólisis (o formación) de un éster, una reacción que la mayoría de los estudiantes encuentran en un curso de laboratorio de química orgánica. Se trata de un proceso más complicado que involucra cinco pasos; su mecanismo se discute aquí. Ver también este sitio de U. Calgary, que describe tanto la reacción catalizada por ácido como base.
Catálisis de oxidación-reducción
Muchas reacciones de oxidación-reducción (transferencia de electrones), incluida la oxidación directa por oxígeno molecular, son bastante lentas. Los iones de metales de transición capaces de existir en dos estados de oxidación a menudo pueden aumentar materialmente la velocidad. Un ejemplo sería la reducción de Fe 3+ por el ion vanadio V 3+:
\[\ce{V^{3+} + Fe^{3+} → V^{4+} + Fe^{2+}}\]
Esta reacción es catalizada por Cu + o Cu 3+, y la velocidad es proporcional a la concentración de V 3+ y del ion cobre, pero independiente de la concentración de Fe 3+. Se cree que el mecanismo implica dos pasos:
| 1 V 3+ + Cu 2+ |
(determinación de la tasa) |
| 2 Fe 3+ + Cu + |
(muy rápido) |
(Si se usa Cu + como catalizador, primero se oxida a Cu 2+ en la etapa 2.)
Peróxido de hidrógeno, de nuevo
Los iones capaces de ser oxidados por un agente oxidante como H 2 O 2 pueden servir como catalizadores para su descomposición. Así, H 2 O 2 oxida el ion yoduro a yodato, cuando luego reduce otra molécula H 2 O 2, devolviendo un ión I — para iniciar el ciclo de nuevo:
\[H_2O_2 + I^– → H_2O + IO^–\]
\[H_2O_2 + IO^– → H_2O + O_2 + I^–\]
El hierro (II) puede hacer lo mismo. Incluso trazas de hierro metálico pueden producir suficiente Fe 2+ para descomponer soluciones de peróxido de hidrógeno.
\[H_2O_2 + Fe^{2+} → H_2O + Fe^{3+}\]
\[H_2O_2 + Fe^{3+} + 2H^+ → H_2O + O_2 + Fe^{2+} + 2H^+\]
Catalizadores heterogéneos
Como su nombre lo indica, existe un catalizador heterogéneo como una fase separada (casi siempre un sólido) de aquella (más comúnmente un gas) en la que tiene lugar la reacción. El efecto catalítico surge de la alteración (a menudo conduce a la disociación) de las moléculas reaccionantes provocadas por su interacción con la superficie del catalizador.
← Modelo de un catalizador que consiste en racimos de 8-10 átomos de platino (azul) depositados sobre una superficie de óxido de aluminio. Este catalizador elimina eficientemente los átomos de hidrógeno del propano, convirtiéndolo en el propileno industrialmente importante. [fuente]
Fuerzas desequilibradas en las superficies
Recordarás que una propiedad universal de la materia son las débiles fuerzas atractivas que surgen cuando dos partículas se acercan de cerca entre sí. (Ver aquí para una revisión rápida.) Cuando las partículas tienen cargas eléctricas opuestas o entran en enlace covalente, estas atracción mucho más fuerte dominan y definen la “química” de las especies que interactúan.
Las unidades moleculares dentro del grueso de un sólido están unidas a sus vecinos a través de estas fuerzas que actúan en direcciones opuestas para mantener cada una bajo una especie de “tensión” que restringe su movimiento y contribuye a la cohesión y rigidez del sólido.
En la superficie de cualquier tipo de materia condensada, las cosas son bastante diferentes. Los átomos o moléculas que residen en la superficie experimentan fuerzas desequilibradas que les impiden asumir las mismas energías de bajo potencial que caracterizan a las unidades interiores. (Lo mismo sucede en los líquidos, y da lugar a una variedad de efectos interfaciales como la tensión superficial).
Pero en el caso de un sólido, en el que las fuerzas atractivas tienden a ser más fuertes, sucede algo mucho más significativo. Las unidades moleculares que residen en la superficie pueden considerarse parcialmente enterradas en ella, con sus partes sobresalientes (y las atracciones intermoleculares que emergen de ellas) expuestas al mundo exterior. La fuerza del campo de fuerza atractivo que emana de una superficie sólida varía en fuerza dependiendo de la naturaleza de los átomos o moléculas que componen el sólido.
No confunda una sorción con una b sorción; esta última se refiere a la absorción masiva de una sustancia en el interior de un material poroso. A nivel microscópico, por supuesto, la absorción también implica adsorción. El proceso en el que las moléculas en un gas o un líquido entran en contacto y se adhieren a una superficie sólida se conoce como adsorción. La adsorción es casi siempre un proceso exotérmico y su resistencia se expresa convencionalmente por la entalpía o “calor” de adsorción Δ H ads.
Quimisorción y Fisisorción
Se reconocen comúnmente dos categorías generales de adsorción, dependiendo de la medida en que se vea afectada la estructura electrónica o de unión de la molécula unida. Cuando las fuerzas atractivas surgen de interacciones relativamente débiles de van der Waals, hay poco efecto y los anuncios Δ H tienden a ser pequeños. Esta condición se describe como adsorción física (fisisorción). La fisisorción de un gas a una superficie es energéticamente similar a la condensación del gas a un líquido, generalmente acumula múltiples capas de moléculas adsorbidas y procede con energía de activación cero.
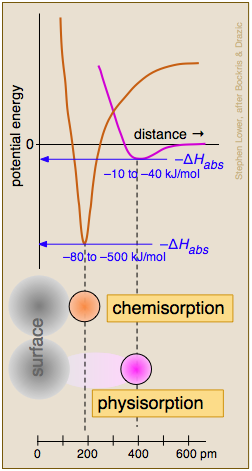
De mayor relevancia para los fenómenos catalíticos es la quimisorción, en la que el adsorbato se une a la superficie por lo que equivale a un enlace químico. La alteración resultante de la estructura electrónica de las especies adsorbidas la “activa” y la hace susceptible a una reacción química (a menudo disociación) que no se podría lograr fácilmente a través de la activación térmica en la fase gaseosa o líquida. En contraste con la fisisorción, la quimisorción generalmente implica una energía de activación (suministrada por Δ H ads) y la especie adornada es siempre una monocapa.
Mecanismos de reacciones en superficies
Adsorción disociativa
El proceso heterogéneo más simple es la quimisorción seguida de la ruptura de enlaces como se describió anteriormente. El más común y minuciosamente estudiado de estos es la disociación del hidrógeno que tiene lugar en la superficie de la mayoría de los metales de transición. El electrón único de 1 s de cada átomo de hidrógeno se coordina con los orbitales d del metal, formando un par de enlaces de quimiosorción (indicados por las líneas discontinuas rojas). Aunque estos nuevos enlaces son más estables que el enlace covalente simple que reemplazan, los átomos de hidrógeno resultantes son capaces de migrar a lo largo de la superficie debido a la extensión continua de la banda de conducción d -orbital.
El mecanismo Langmuir-Hinshelwood
Aunque los átomos adsorbidos (” adatomos “) no son radicales libres, sin embargo son altamente reactivos, por lo que si una segunda especie molecular diferente se adsorbe sobre la misma superficie, puede ser posible un intercambio de átomos. Así, el monóxido de carbono puede oxidarse a CO 2 mediante el procedimiento ilustrado a continuación:
En este ejemplo, solo la molécula O 2 sufre disociación. La molécula de CO se adsorbe sin disociación
![]() , configurada perpendicular a la superficie con el enlace de quimiosorción centrado sobre un espacio hueco entre los átomos metálicos. Después de que las dos especies adsorbidas hayan migrado una cerca de la otra
, configurada perpendicular a la superficie con el enlace de quimiosorción centrado sobre un espacio hueco entre los átomos metálicos. Después de que las dos especies adsorbidas hayan migrado una cerca de la otra, el átomo de oxígeno cambia su unión de la superficie metálica para formar un enlace C=O más estable con el carbono
, seguido de la liberación de la molécula del producto.
El mecanismo Eley-Rideal
Un mecanismo alternativo elimina la segunda etapa de quimisorción; los adatomeos de oxígeno reaccionan directamente con las moléculas de CO gaseosas reemplazando el enlace de quimiosorción por un nuevo enlace C-O a medida que se abalanzan sobre la superficie:
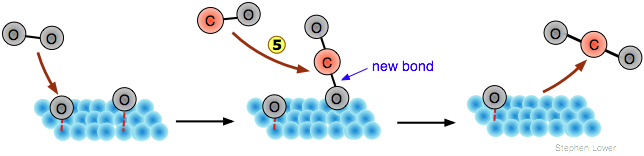
Se conocen ejemplos de ambos mecanismos, pero el mecanismo Langmuir-Hinshelwood es más importante que explota la activación del reactivo adsorbido. En el caso de la oxdación de monóxido de carbono, los estudios que involucran experimentos de haz molecular respaldan este esquema. Una evidencia clave es la observación de un breve lapso de tiempo entre el contacto de una molécula de CO con la superficie y la liberación del CO 2, lo que sugiere que el CO permanece quimisorbido durante el intervalo.
El principio de Sabatier
Para ser efectivos, estos procesos de adsorción, reacción y desorción deben ser orquestados de una manera que dependa críticamente de las propiedades del catalizador en relación con las propiedades de quimiosorción (Δ H ads) de los reactivos y productos.
- La adsorción del reactivo sobre la superficie catalítica (2) debe ser lo suficientemente fuerte como para perturbar la unión dentro de la especie para disociarla o activarla;
- Si los fragmentos resultantes deben migrar a otros lugares de la superficie (3-4), su quimisorción debe ser lo suficientemente débil como para permitir este movimiento pero no tan pequeña como para que escapen antes de que tengan la oportunidad de reaccionar;
- La especie de producto debe tener valores de anuncios Δ H suficientemente pequeños para asegurar su rápida desorción del catalizador (5) de manera que la superficie se libere para repetir el ciclo
La importancia de elegir un catalizador que logre el equilibrio adecuado de los calores de adsorción de los diversos componentes de la reacción se conoce como el Principio Sabatier, pero a veces se le conoce como el principio de “just-right” o “Ricitos de Oro”. ¿Recuerdas la historia de Ricitos de Oro y los Tres Osos? ... o ver este video de uTube.
En su aplicación a la catálisis, este principio se ilustra frecuentemente mediante un “diagrama volcánico” en el que la velocidad de la reacción catalizada se representa en función de los anuncios Δ H de un sustrato como H 2 sobre una superficie de metal de transición.
La gráfica de la izquierda muestra la efectividad relativa de varios metales en la catalización de la descomposición del ácido fórmico HCOOH. El eje vertical se traza como temperatura, siendo la idea que cuanto mejor sea el catalizador, menor será la temperatura requerida para mantener una tasa dada.
El ciclo catalítico
Este término se refiere a la secuencia idealizada de etapas entre la adsorción de un reactivo sobre el catalizador y la desorción del producto, culminando en la restauración del catalizador a su condición original. A continuación se ilustra un ciclo catalítico típico para la hidrogenación de propeno.
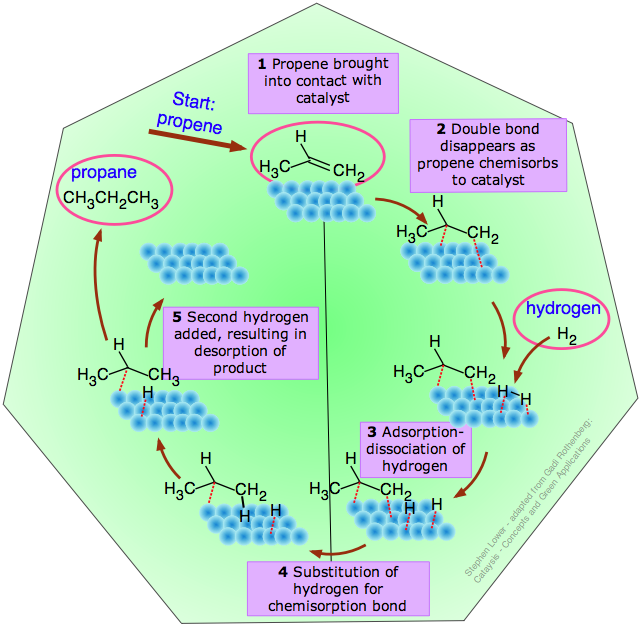
Esta reacción particular
H 3 C—CH=CH 2 + H 2 → H 3 C—CH 2 —CH 3
se lleva a cabo espontáneamente sólo en sentido inverso, pero es representativo del proceso utilizado para hidrogenar los dobles enlaces carbono-carbono en los aceites vegetales para producir grasas sólidas saturadas como la margarina.
Intoxicación y descomposición del catalizador
El envenenamiento por catalizadores, provocado por la unión irreversible de una sustancia a su superficie, puede ser permanente o temporal. En este último caso el catalizador se puede regenerar, generalmente calentando a una temperatura alta. En los organismos, muchas de las sustancias que conocemos como “venenos” actúan como venenos catalíticos sobre las enzimas. Si los catalizadores realmente permanecen sin cambios, deberían durar para siempre, pero en la práctica real, pueden ocurrir diversos eventos que limitan la vida útil de muchos catalizadores.
- Las impurezas en la materia prima o los productos de reacciones secundarias pueden unirse permanentemente a un número suficiente de sitios activos para reducir la eficiencia catalítica con el tiempo, “envenenando” el catalizador.
- El deterioro físico del catalizador o de su soporte, a menudo provocado por las altas temperaturas a veces utilizadas en los procesos industriales, puede reducir la superficie efectiva o la accesibilidad de los reactivos a los sitios activos.
Los catalizadores tienden a ser bastante caros, por lo que es ventajoso si pueden ser reprocesados o regenerados para restaurar su actividad. Es una práctica industrial común cerrar periódicamente las unidades de proceso para reemplazar los catalizadores gastados.
Cómo funcionan los catalizadores heterogéneos
Los mecanismos reales por los cuales la adsorción de una molécula sobre una superficie catalítica facilita la escisión de un enlace varían mucho de un caso a otro.
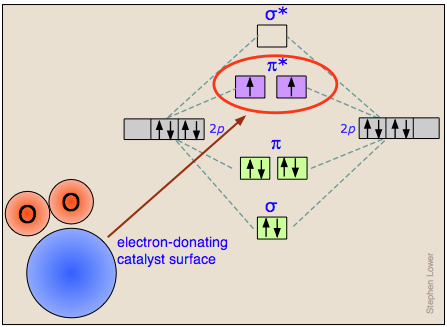
Damos aquí solo un ejemplo, el de la disociación del dixógeno O 2 en la superficie de un catalizador capaz de donar temporalmente un electrón que entra en un orbital molecular antienlace de oxígeno que desestabilizará claramente el enlace O—O. (Una vez que se ha roto el enlace, el electrón se devuelve al catalizador).
Tipos de superficies catalíticamente activas
Los catalizadores heterogéneos dependen principalmente de uno o más de los siguientes tipos de superficies:
| Tipo de superficie activa | Observaciones |
|---|---|
| Los átomos en las superficies o bordes de los cristales en redes macromoleculares 2 y 3-D como el grafito o el cuarzo pueden tener valencias libres | |
| Las superficies y los bordes son sitios de campos eléctricos intensos capaces de interactuar con iones y moléculas polares | |
| Los óxidos pueden adquirir grupos H + y/o OH — capaces de actuar como catalizadores ácidos o básicos | |
| El “gas de electrones” en las superficies metálicas puede perturbar la unión en las moléculas de sustrato | |
 |
Los orbitales d vacantes pueden proporcionar una variedad de sitios de coordinación para la activación. |
| Los semiconductores (incluyendo muchos óxidos) pueden suministrar electrones, excitados térmicamente a través de brechas de banda razonablemente pequeñas (<50 kJ). |
Algunos factores que afectan la eficacia del catalizador
Dado que la catálisis heterogénea requiere el contacto directo entre los reactivos y la superficie catalítica, el área de superficie activa va en la parte superior de la lista. En el caso de una película metálica, esto no es lo mismo que el área nominal de la película medida por una regla; a nivel microscópico, incluso las superficies aparentemente lisas son altamente irregulares, y algunas cavidades pueden ser demasiado pequeñas para acomodar moléculas reaccionantes.
Considere, por ejemplo, que un cubo de platino de 1 cm (que cuesta aproximadamente $1000) tiene una superficie nominal de solo 6 cm 2. Si esto se descompone en 10 12 cubos más pequeños cuyos lados son de 10 —6 m, la superficie total sería de 60,000 cm 2, capaces en principio de incrementar la velocidad de una reacción catalizada por PT en un factor de 10 4. Estos metales muy finamente divididos (y a menudo muy caros) se unen típicamente a una superficie de soporte inerte para maximizar su exposición a los reactivos.
Topografía superficial. A nivel microscópico, incluso una superficie aparentemente lisa está deshuesada y desigual, y algunos sitios serán más activos que otros. La penetración de moléculas dentro y fuera de algunos de los canales más pequeños de una superficie porosa puede llegar a ser limitante de la velocidad.
Una superficie lisa siempre poseerá una variedad de defectos como escalones y esquinas que ofrecen una mayor exposición y pueden ser los únicos sitios activos en la superficie, o demasiado activos para unirse permanentemente a un reactivo, reduciendo el área activa de la superficie. En un estudio, se determinó que los defectos de torcedura que constituyen apenas el 5 por ciento de la superficie del platino fueron responsables de más del 90% de la actividad catalítica en una determinada reacción.
Factores estéricos
Cuando la quimisorbción ocurre en dos o más ubicaciones en el reactivo, la catálisis eficiente requiere que la separación de los centros activos en la superficie catalítica sea tal que se puedan formar enlaces superficiales sin distorsión angular significativa.
Así, la activación del doble enlace etileno sobre una superficie de níquel procede de manera eficiente porque el ángulo entre los enlaces C—Ni y el C-C está cerca del valor tetraédrico de 109.5° requerido para la formación orbital híbrida de carbono sp 3. De igual manera, podemos esperar que la hidrogenación del benceno proceda de manera eficiente sobre una superficie en la que los sitios activos estén espaciados en el rango de 150 y 250pm.
Esta es una razón por la que muchos catalizadores metálicos exhiben diferente actividad catalítica en diferentes caras cristalinas.
Algunos tipos especiales de catalizadores heterogéneos
Catalizadores de conglomerados metálicos
A medida que se reduce el tamaño de partícula de un catalizador, aumenta la fracción de átomos de escalón, borde y esquina más expuestos. Un caso extremo ocurre con estructuras de conglomerados metálicos de tamaño nanométrico (1-2 nm) compuestas típicamente de 10-50 átomos.
[enlace] →
El oro metálico, bien conocido por su inercia química, exhibe una actividad catalítica muy alta cuando se deposita como cúmulos metálicos sobre un soporte de óxido. Por ejemplo, O 2 se disocia fácilmente en agrupaciones de Au 55 que se ha encontrado que catalizan eficientemente la oxidación de hidrocarburos [artículo].
Catalizadores de zeolita
Las zeolitas son sólidos de aluminosilicato tipo arcilla que forman estructuras microporosas de marco abierto que pueden contener jaulas, cavidades o canales unidos cuyas dimensiones se pueden adaptar a los tamaños de los reactivos y productos. Para aquellas moléculas capaces de difundirse a través de estos espacios, las zeolitas son en efecto “toda la superficie”, haciéndolas altamente eficientes. Esta selectividad por tamaño los hace importantes para aplicaciones de adsorción, separación, intercambio iónico y catalíticas. Muchas zeolitas se presentan como minerales, pero otras se elaboran sintéticamente con el fin de optimizar sus propiedades.
Como catalizadores, las zeolitas ofrecen una serie de ventajas que las han hecho especialmente importantes en operaciones de “química verde” en las que se minimiza el número de etapas de procesamiento, subproductos no deseados y volúmenes de corrientes residuales.
Enzimas y biocatálisis
“Una Jarra de Vino, una Hogaza de Pan, y.”. ¡Enzimas!
Esta distorsión de la ya distorsionada traducción de Robert FitzGerald de la famosa cuarteta del maravilloso Rubaiyat de Omar Khayyam subraya el papel central que las enzimas y su tecnología han jugado en la civilización desde la antigüedad.
← Ilustración por Edmund Dulac (1882-1953)
La fermentación y la vinificación han sido parte de la historia y la cultura humanas desde hace al menos 8000 años, pero el reconocimiento del papel de la catálisis en estos procesos tuvo que esperar hasta finales del siglo XIX. Para la década de 1830, se habían descubierto numerosos agentes similares, como los que facilitan la digestión de proteínas en el estómago. El término “enzima”, que significa “de levadura”, fue acuñado por el fisiólogo alemán Wilhelm Kühne en 1876. En 1900, Eduard Buchner (1860-1917, 1907 Premio Nobel de Química) demostró que la fermentación, que antes se creía dependía de una misteriosa “fuerza vital” contenida en organismos vivos como la levadura, se podía lograr mediante un “jugo de prensa” libre de células que exprimió de la levadura.
Para entonces se reconoció que las enzimas son una forma de catalizador (término introducido por Berzelius en 1835), pero su naturaleza química exacta quedó en duda. Parecían estar asociados con proteínas, pero la comprensión general de que las enzimas son proteínas comenzó solo en la década de 1930 cuando se cristalizó la primera enzima pura, y no llegó a ser generalmente aceptada hasta la década de 1950. Ahora está claro que casi todas las enzimas son proteínas, siendo la principal excepción una pequeña pero importante clase de enzimas basadas en ARN conocidas como ribozimas.
Las proteínas están compuestas por largas secuencias de aminoácidos encadenados entre sí por enlaces amida; esta secuencia define la estructura primaria de la proteína. Su enorme tamaño (típicamente 200-2000 unidades de aminoácidos, con pesos moleculares totales 20,000 - 200,000) les permite plegarse de formas complicadas (conocidas como estructuras secundarias y terciarias) cuyas configuraciones son esenciales para su función catalítica.
Debido a que las enzimas son generalmente mucho más grandes que las moléculas reaccionantes sobre las que actúan (conocidas en bioquímica como sustratos), la catálisis enzimática es de alguna manera similar a la catálisis heterogénea. La principal diferencia es que la unión de un subtrato a la enzima es mucho más selectiva.
Precursores y cofactores
La mayoría de las enzimas vienen a existir como precursores inactivos (zimógenos) que se convierten a sus formas activas en el momento y lugar en que se necesitan.
- Por ejemplo, se supone que las enzimas que conducen a la coagulación de la sangre permanecen inactivas hasta que realmente comience el sangrado; un factor activador importante es la exposición de la sangre a proteínas en la pared del vaso dañado.
- La enzima que cataliza la formación de lactosa (azúcar de la leche) en la glándula mamaria se forma durante el embarazo, pero permanece inactiva hasta el momento del nacimiento, cuando los cambios hormonales hacen que una unidad modificadora se una y active la enzima.
La conversión a la forma activa puede implicar una simple ruptura de la proteína por hidrólisis de un enlace peptídico apropiado o la adición de un fosfato o grupo similar a uno de los residuos de aminoácidos.
Muchas proteínas enzimáticas también requieren moléculas “colaboradoras”, conocidas como cofactores, para hacerlas catalíticamente activas. Estos pueden ser iones metálicos simples (muchos de los iones nutritivos traza de Cu, Mn, Mo, V, etc.) o pueden ser moléculas orgánicas más complejas que se denominan coenzimas. Muchos de estos últimos son lo que comúnmente denominamos vitaminas. Otras moléculas, conocidas como inhibidores, disminuyen la actividad enzimática; muchas drogas y venenos actúan de esta manera.
El complejo enzima-sustrato
El modelo estándar de cinética enzimática consiste en un proceso de dos etapas en el que una enzima se une reversiblemente a su sustrato S (el reactivo) para formar un complejo enzima-sustrato ES:
El complejo enzima-sustrato juega un papel similar al del complejo activado en la cinética convencional, pero la función principal de la enzima es estabilizar el estado de transición.
En la segunda etapa, esencialmente irreversible, se liberan el producto y la enzima:
El tratamiento cinético básico de este proceso implica la suposición de que las concentraciones [E] y [ES] alcanzan valores de estado estacionario que no cambian con el tiempo. (El tratamiento detallado, que está más allá del alcance de este curso, se puede encontrar aquí.)
El proceso general es descrito por la ecuación de Michaelis-Menten que se traza aquí. La constante de Michaelis k M se define como se muestra, pero puede simplificarse a la constante de disociación ES k —1/k 1 en los casos en que la disociación del complejo es el paso limitante de la velocidad. La cantidad v max no se observa directamente, pero se puede determinar a partir de k M como se muestra aquí.
Las enzimas son proteínas
Para comprender las enzimas y cómo catalizan las reacciones, primero es necesario revisar algunos conceptos básicos relacionados con las proteínas y los aminoácidos de los que están compuestas.
Aminoácidos: polares, no polares, positivos, negativos.
Los 21 aminoácidos que componen las proteínas poseen la estructura básica aquí mostrada, donde R representa hidrógeno o una cadena lateral que puede contener grupos -NH 2 o -COOH adicionales. Ambos tipos de grupos pueden formar enlaces de hidrógeno con el agua y con las partes polares de los sustratos, y por lo tanto contribuyen a la polaridad de los aminoácidos y a la naturaleza hidófila. Las cadenas laterales que contienen cadenas de carbono más largas y especialmente anillos de benceno tienen el efecto contrario, y tienden a hacer que el aminoácido sea no polar e hidrófobo.
Tanto los grupos -NH 2 como —COOH son ionizables (es decir, pueden actuar como donantes o aceptores de protones) y cuando están en sus formas iónicas, tendrán una carga eléctrica. Los grupos —COOH tienen pKa en el rango de 1.8-2.8, y por lo tanto estarán en sus formas ionizadas —COO — a valores de pH celulares ordinarios de alrededor de 7.4. Los pKa del grupo amino están alrededor de 8.8-10.6, por lo que estos también estarán normalmente en sus formas ionizadas NH 3 +.
Esto significa que a pH celular ordinario, tanto los grupos carboxilo como amino serán ionizados. Pero debido a que las cargas tienen signos opuestos, un aminoácido que no tenga grupos ionizables adicionales en su cadena lateral tendrá una carga neta de cero. Pero si la cadena lateral contiene un grupo amino o carboxilo extra, el aminoácido puede llevar una carga eléctrica neta. El siguiente diagrama ilustra aminoácidos típicos que caen dentro de cada uno de los grupos descritos anteriormente.
Proteínas
Las proteínas se componen de una o más cadenas de aminoácidos unidas entre sí a través de enlaces peptídicos mediante la eliminación de una molécula de agua.
El producto mostrado anteriormente se llama péptido, específicamente es un dipéptido porque contiene dos residuos de aminoácidos (lo que queda después de que se haya eliminado el agua). Las proteínas son simplemente cadenas polipeptídicas muy largas, o combinaciones de ellas. (¡La distinción entre un polipéptido largo y una proteína pequeña es bastante borrosa!)
Proteínas globulares
La mayoría de las enzimas entran en la categoría de proteínas globulares. A diferencia de las proteínas fibrosas que forman los componentes estructurales de los tejidos, las proteínas globulares son solubles en agua y rara vez tienen estructuras terciarias sistemáticas. Están formados por una o más cadenas de aminoácidos (” péptido “) que se pliegan en varias formas que pueden describirse aproximadamente como esféricas, de ahí el término “globular”, y el sufijo “globina” que frecuentemente se agrega a sus nombres, como en “hemoglobina”.
El plegamiento proteico es un proceso espontáneo que está influenciado por una serie de factores. Una de ellas es obviamente su secuencia primaria de aminoácidos que facilita la formación de enlaces intramoleculares entre aminoácidos en diferentes partes de la cadena. Estos consisten principalmente en enlaces de hidrógeno, aunque los enlaces disulfuro S-S entre aminoácidos que contienen azufre no son infrecuentes.
Además de estas fuerzas intramoleculares, las interacciones con el entorno juegan un papel importante. Lo más importante de estos es el efecto hidrofóbico, que favorece conformaciones de plegamiento en las que los aminoácidos polares (que forman enlaces de hidrógeno con el agua) están en el exterior, mientras que los llamados aminoácidos hidrófobos permanecen en ubicaciones protegidas dentro de los pliegues.
Cómo funcionan las enzimas: el sitio activo
El proceso catalítico mediado por una enzima tiene lugar en una depresión o hendidura que expone el sustrato a solo unos pocos de los cientos a miles de residuos de aminoácidos en la cadena proteica. La alta especificidad y actividad de la catálisis enzimática depende sensiblemente de la forma de esta cavidad y de las propiedades de los aminoácidos circundantes
En 1894, mucho antes de que fuera claro que las enzimas son proteínas, el químico alemán Emil Fischer sugirió el llamado modelo de bloqueo y llave como una forma de entender cómo una enzima dada puede actuar de manera específica sobre un solo tipo de molécula sustrato. Este modelo es esencialmente una elaboración del que todavía usamos para explicar catálisis heterogénea.
Aunque el modelo básico de bloqueo y llave sigue siendo útil, se ha modificado en lo que ahora se llama el modelo de ajuste inducido. Esto supone que cuando el sustrato ingresa al sitio activo e interactúa con las partes circundantes de la cadena de aminoácidos, reforma el sitio activo (y quizás otras partes de la enzima) para que pueda engancharse más completamente con el sustrato.
Un paso importante en este proceso es exprimir cualquier molécula de agua que esté unida al sustrato y que interfiera con su posicionamiento óptimo.
Dentro del sitio activo, las interacciones específicas entre el sustrato y los aminoácidos apropiadamente cargados, hidrófilos e hidrófobos del sitio activo estabilizan entonces el estado de transición distorsionando la molécula sustrato de tal manera que conduzca a un estado de transición que tiene una activación sustancialmente menor. energía que se puede lograr mediante catálisis ordinay no enzimática. Más allá de este punto, las etapas catalíticas básicas son bastante convencionales, siendo la catálisis ácido/base y nucleofílica la más común.
Para una ilustración muy clara e instructiva de una secuencia típica de varios pasos de una reacción típica catalizada por enzimas, vea esta página del libro de texto en línea de Mark Bishop, del que se toma esta ilustración.
Regulación e inhibición de enzimas
Si todas las enzimas de un organismo estuvieran activas todo el tiempo, el resultado sería un caos desbocado. La mayoría de los procesos celulares, como la producción y utilización de energía, la división celular y la descomposición de los productos metabólicos, deben operar de una manera exquisitamente coreografiada y afinada, al igual que una gran orquesta sinfónica; ¡aquí no hay lugar para la improvisación de jazz!
La naturaleza ha ideado diversas formas de lograrlo; describimos la acción de precursores y coenzimas anteriormente. Aquí nos enfocamos en uno de los mecanismos regulatorios más importantes (y químicamente interesantes).
Regulación alostérica: ajustar el sitio activo
Existe una clase importante de enzimas que poseen sitios especiales (distintos de los sitios catalíticamente activos) a los que ciertas moléculas externas pueden unirse reversiblemente. Si bien estos sitios alostéricos, como se les llama, pueden estar bastante alejados de los sitios catalíticos, el efecto de la unión o liberación de estas moléculas es desencadenar un cambio rápido en el patrón de plegamiento de la enzima que altera la forma del sitio activo. El efecto es permitir que una molécula de señalización o reguladora (a menudo una muy pequeña como el NO) modula la actividad catalítica del sitio activo, activando o apagando efectivamente la enzima.
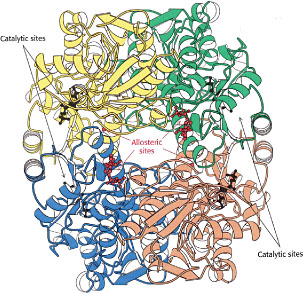
En algunos casos, el producto de una reacción catalizada por enzimas puede unirse a un sitio alostérico, disminuyendo la actividad de la enzima y proporcionando así retroalimentación negativa que ayuda a mantener el producto en la concentración deseada. Se cree que las concentraciones de iones plasmáticos como el calcio, y de ATP que suministra energía, están reguladas de esta manera.
Las enzimas alostéricas son más que catalizadores: actúan como puntos de control en las redes de señalización metabólica y celular.Las enzimas alostéricas frecuentemente se encuentran al comienzo de una secuencia de enzimas en una cadena metabólica, o en puntos de ramificación donde dos de esas cadenas divergen, actuando muy parecido a las señales de tráfico en intersecciones congestionadas.
Inhibición enzimática
Como es el caso de los catalizadores heterogéneos, ciertas moléculas distintas del sustrato normal pueden entrar y ser retenidas en el sitio activo para inhibir competitivamente la actividad de una enzima. Es así como funcionan la penicilina y antibióticos relacionados; estas moléculas se unen covalentemente a residuos de aminoácidos en el sitio activo de una enzima que cataliza la formación de un componente esencial de las paredes celulares bacterianas. Cuando la célula se divide, la progenie recién formada esencialmente se desmorona.
Enzimas fuera de la célula
Las enzimas se han empleado ampliamente en las industrias de alimentos, pulpa y papel y detergentes durante mucho tiempo, pero principalmente como extractos impuros de células completas.
En los últimos años, los desarrollos en biotecnología y el paso gradual de la industria de depender de materias primas y solventes a base de petróleo a la llamada química “verde” han hecho que las enzimas sean más atractivas como catalizadores industriales. En comparación con este último, las enzimas purificadas tienden a ser caras, difíciles de reciclar e inestables fuera de rangos bastante estrechos de temperatura, pH y composición de solventes.
Enzimas inmovilizadas
Muchos de los problemas relacionados con el uso de enzimas libres se pueden superar inmovilizando la enzima. Esto se puede lograr de varias maneras:
- Unión de la enzima a un material de soporte sólido inerte
Esto se hizo por primera vez en 1916, utilizando carbón vegetal o hidróxido de aluminio. desde la década de 1960, cuando este método se hizo más popular, los polímeros sintéticos, a menudo diseñados para una aplicación específica, han entrado en amplio uso. También se han empleado algunos biopolímeros naturales como la celulosa o el almidón, y sólidos inorgánicos como la sílice y la alúmina.
- Atrapamiento de la enzima en un material poroso
- Enzimas reticuladas
Catálisis
- Gady Rothenberg- Catálisis: Conceptos y Aplicaciones Verdes (Wiley-VCH, 2008)
- R.A. van Santan, M. Neurock - Catálisis Molecular Heterogénea: Un enfoque conceptual y computacional (Wiley-VCH, 2006)
- Historia de la Catálisis: Lista de páginas web, incluyendo Cincuenta años de Catálisis - Una lista de los principales avances en el campo 1949-1999. Páginas de historia más detalladas del Reino Unido
- Catálisis y sus aplicaciones - un relato muy legible e interesante de las aplicaciones de la catálisis a diversos campos de importancia industrial.