
17.5: Cinética de reacciones en solución
- Page ID
- 70621

Asegúrese de comprender a fondo las siguientes ideas esenciales:
- Describir algunas de las principales diferencias entre la cinética de reacciones en fase gaseosa, en comparación con las de soluciones líquidas.
- ¿Qué papel desempeñan las jaulas solventes en la cinética de la solución?
- Explicar la distinción entre el control de difusión y el control de activación de las velocidades de reacción en soluciones.
- ¿Cómo puede la polaridad de un solvente afectar la energía de un mecanismo de reacción?
Los fundamentos cinéticos que cubrimos en las secciones anteriores de este grupo de lección se relacionan con procesos que tienen lugar en la fase gaseosa. Pero los químicos y bioquímicos generalmente están mucho más preocupados por las soluciones. Esta lección te llevará a través de algunas de las extensiones de cinética básica que necesitas para entender los principales cambios que ocurren cuando las reacciones ocurren en soluciones líquidas.
¿Qué diferencia tiene la cinética en las soluciones líquidas?
La mayoría de las complicaciones añadidas de la cinética y los procesos de velocidad en soluciones líquidas surgen de la densidad mucho mayor de la fase líquida. En un gas típico a presión atmosférica, las moléculas ocupan sólo alrededor de 0.2 por ciento del volumen; el otro 99.8 por ciento es espacio vacío. En un líquido, las moléculas pueden ocupar más de la mitad del volumen, y los espacios “vacíos” son irregulares y siempre cambiantes a medida que las moléculas solventes experimentan movimientos térmicos propios.

En una solución líquida típica, las moléculas de disolvente superan masivamente en número a las moléculas de soluto reactivo, que tienden a encontrarse momentáneamente (~10-11 segundos) confinadas a un “agujero” dentro del líquido. Este atrapamiento se vuelve especialmente importante cuando el disolvente está fuertemente unido a hidrógeno como es el caso del agua o el alcohol.

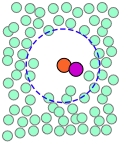
Cuando los movimientos térmicos ocasionalmente liberan una molécula de soluto de esta trampa, saltará a una nueva ubicación. Los saltos son muy rápidos (10 —12 - 10 —13 seg) y cortos (generalmente unos pocos diámetros de disolvente-molécula), y siguen un patrón completamente aleatorio, muy parecido al movimiento browniano. Considera un proceso bimolecular sencillo A + B → productos. Las moléculas reaccionantes generalmente saltarán de un agujero a otro en la matriz solvente, solo ocasionalmente se encontrarán en la misma jaula de solventes donde es probable que los movimientos térmicos las pongan en contacto.
 |
 |
 |
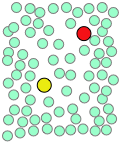 |
| Un par de reactivos terminan en la misma jaula de solventes, donde rebotan aleatoriamente e intercambian energía cinética con las moléculas de solvente. | Eventualmente los dos reactivos forman un par de encuentro. Si no logran reaccionar la primera vez, tienen muchas más oportunidades durante la vida de la jaula. | Los productos se forman y comienzan a alejarse unos de otros. | Finalmente, después de aproximadamente 10 —11 s, la jaula del solvente se rompe y los productos se difunden. |
El proceso se puede representar como
\[A + B \rightarrow \{AB\} → \text{products}\]
en el que el\(\{AB\}\) término representa los reactivos enjaulados incluyendo el par de encuentro y el complejo activado.
Contraste este escenario con una reacción similar que tiene lugar en fase gaseosa; las moléculas involucradas en la reacción a menudo serán las únicas presentes, por lo que una proporción significativa de las colisiones serán\(A\):\(B\) encuentros. Sin embargo, si la colisión no fuera energética o geométricamente viable, las moléculas reaccionantes se separan volando y es poco probable que vuelvan a encontrarse pronto. En un líquido, sin embargo, las moléculas de soluto están efectivamente en un estado constante de colisión, si no con otros reactivos, entonces con moléculas de disolvente que pueden intercambiar energía cinética con los reactivos. Entonces, una vez que se forma un par de encuentros A-B, los dos reactivos reciben múltiples golpes el uno al otro, aumentando enormemente la probabilidad de que obtengan la energía cinética necesaria para patearlos sobre la joroba de activación antes de que la pareja de encuentro se desintegre.
Casos limitantes: Reacciones controladas por difusión y controladas por activación
El modelo de par de encuentros introduce algunos nuevos parámetros de velocidad:
\[\ce{A + B <=>[k1][k_{-1}] {AB} -> products}\]
El primer paso es un equilibrio entre los reactivos fuera y dentro de la jaula de solventes. Las constantes de velocidad\(k_1\) y\(k_2\) reflejan las relacionadas con la difusión de moléculas a través del disolvente; sus valores son fuertemente dependientes de la viscosidad (y por lo tanto de la temperatura) del disolvente. (Tenga en cuenta que\(k_1\) es una constante de tasa de segundo orden, mientras que\(k_2\) es de primer orden.)
La difusión es el transporte de una sustancia a través de un gradiente de concentración; es decir, de una región de mayor concentración a una de menor concentración. Piense en la forma en que el color del té se extiende cuando una bolsa de té se sumerge en agua caliente. La difusión ocurre porque los movimientos térmicos aleatorios son estadísticamente más propensos a mover moléculas fuera de una región de mayor concentración que en la dirección inversa, simplemente porque en este último caso hay menos moléculas disponibles para hacer el viaje inverso. Eventualmente las concentraciones se vuelven uniformes y se alcanza el equilibrio.
A medida que las moléculas se difunden a través de un líquido, deben apartar a las moléculas vecinas. El trabajo requerido para ello equivale a una energía de activación, por lo que la difusión puede considerarse como un proceso cinético con su propia constante de velocidad k d y energía de activación. Estos parámetros dependen de los tamaños de las moléculas de soluto y disolvente y de la fuerza con la que estas últimas interactúan entre sí. Esto sugiere dos casos limitantes importantes para las reacciones en solución.
Para el agua a temperatura ambiente, k 1 es típicamente 10 9 -10 10 dm —3 mol —1 s —1 y k 2 es alrededor de 10 —9 -10 —10 dm —3 mol —1 s —1 . Dados estos valores, k 3 > 10 12 s —1 implica control de difusión, mientras que los valores < 10 9 s —1 son indicativos de control de activación.
- Difusión Controlada (\(k_3 \gg k_2\)): Si la energía de activación de la reacción A+B es muy pequeña o si el escape de moléculas de la jaula {AB} es difícil, la cinética estará dominada por k 1, y así por la energía de activación de difusión. Se dice que tal proceso está controlado por difusión. Reacciones en solución acuosa en las que es probable que E a > 20 kJ/mol entren en esta categoría.
- Activación controlada (\(k_3 \ll k_2\)): Alternativamente, si la energía de activación de la reacción A+B domina la cinética, y la reacción está controlada por activación.
Varios tipos generales de reacciones son consistentemente muy “rápidas” y, por lo tanto, se encuentran comúnmente controladas por difusión en la mayoría de los disolventes:
Las Unidades Matter
Las constantes de velocidad en fase gaseosa se expresan normalmente en unidades de mol s —1, pero las constantes de velocidad de las reacciones en solución se dan convencionalmente en unidades mol/L, o dm 3 mol —1 s —1. La conversión entre ellos depende de una serie de suposiciones y no es trivial.
- Recombinación de átomos y radicales
Por ejemplo la formación de I 2 a partir de I átomos en hexano a 298 K tiene k 3 = 1.3×10 12 dm 3 mol —1 s —1.
- Las reacciones ácido-base que implican el transporte de iones H + y OH — tienden a ser muy rápidas.
La más famosa de estas es una de las reacciones más rápidas conocidas:\[H^+ + OH^– → H_2O \nonumber \] para la cual k 3 = 1.4×10 11 dm 3 mol —1 s —1 a 298 K.
Los disolventes polares como el agua y los alcoholes interactúan con iones y moléculas polares a través de atractivas interacciones dipolo-dipolo e ión-dipolo, dando lugar a formas solvatadas de menor energía que estabilizan estas especies. De esta manera, un disolvente polar puede alterar tanto la termodinámica como la cinética (velocidad) de una reacción.
Efecto Termodinámico Solvente
Si los productos de la reacción son marcadamente más o menos polares que los reactivos, la polaridad del disolvente puede cambiar la termodinámica general (constante de equilibrio) de la reacción. En ninguna parte esto es más evidente que cuando un sólido iónico como la sal se disuelve en agua. Los iones Na + y Cl — se unen en el sólido a través de fuertes fuerzas culómbicas; separar el sólido en vacío o en un disolvente no polar es un proceso altamente endotérmico. En contraste, la disolución de NaCl en agua es ligeramente exotérmica y procede espontáneamente.
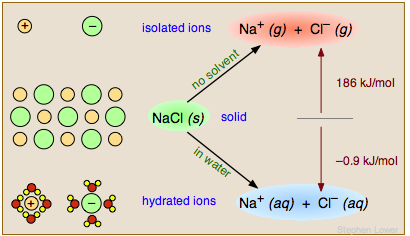
El agua facilita este proceso de dos maneras importantes. Primero, su alta constante dieléctrica de 80 reduce la fuerza entre los iones separados a 1/80 de su valor normal. En segundo lugar, las moléculas de agua forman una capa de solvatación alrededor de los iones (inferior izquierda), volviéndolos energéticamente (termodinámicamente) más estables que en el sólido de NaCl.
Efecto cinético del solvente
De la misma manera, una reacción cuyo mecanismo implique la formación de un complejo intermedio o activado que tenga un carácter polar o iónico tendrá su energía de activación, y así su velocidad, sujeta a cambios a medida que se altere la polaridad del disolvente. Como ejemplo consideraremos una clase importante de reacciones de las que escucharás mucho si tomas un curso de química orgánica. Cuando se añade una solución acuosa de una base fuerte tal como KOH a una solución de cloruro de butilo terciario en etanol, el cloro se reemplaza por un grupo hidroxilo, dejando alcohol t-butílico como producto:
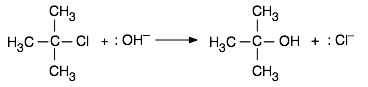
Esta reacción es una de una clase grande e importante conocida como procesos de sustitución nucleofílica S N 1 que se discuten en la mayoría de los cursos de química orgánica. En estas reacciones, una especie que posee un par de electrones no enlazantes (también llamados nucleófilos o base de Lewis) los utiliza para formar un nuevo enlace con un electrófilo, un compuesto en el que un átomo de carbono tiene una carga positiva parcial debido a sus enlaces a electrones- retirando grupos. En el ejemplo aquí, otros nucleófilos como NH 3 o incluso H 2 O servirían también.
Para reflejar la generalidad de este proceso y enfocarnos en los grandes cambios que se producen, representaremos esta reacción como
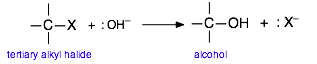
Estudios extensos de esta clase de reacciones en la década de 1930 revelaron que procede en dos etapas de activación controlada por energía, seguidas de una simple disociación en los productos:

En el paso , que es determinante de la velocidad, el cloro sale del cloruro de alquilo que se convierte en un intermedio conocido como carbocatión (“ion gato”). Estos iones, en los que el átomo de carbono central carece de un octeto completo, son altamente reactivos, y en escalón
, que es determinante de la velocidad, el cloro sale del cloruro de alquilo que se convierte en un intermedio conocido como carbocatión (“ion gato”). Estos iones, en los que el átomo de carbono central carece de un octeto completo, son altamente reactivos, y en escalón el carbocatión es atacado por el ion hidróxido que suministra el electrón faltante. El producto inmediato es otro catión en el que la carga positiva está en el átomo de oxígeno. Este ion oxonio es inestable y se disocia rápidamente (
el carbocatión es atacado por el ion hidróxido que suministra el electrón faltante. El producto inmediato es otro catión en el que la carga positiva está en el átomo de oxígeno. Este ion oxonio es inestable y se disocia rápidamente ( ) en el alcohol y un ion hidrógeno.
) en el alcohol y un ion hidrógeno.

El diagrama de coordenadas de la reacción nos ayuda a comprender el efecto de la polaridad del disolvente en esta reacción. Las moléculas de disolvente polar interactúan más fuertemente con especies en las que la carga eléctrica se concentra en un solo punto. Así, el carbocatión se estabiliza en mayor grado que los complejos activados en los que la carga se extiende entre los extremos positivo y negativo. Como indican las flechas verdes pesadas, un disolvente más polar estabilizará el carbocatión más que cualquiera de los complejos activados; el efecto es reducir materialmente la energía de activación de la etapa determinante de la velocidad, y así acelerar la reacción. Debido a que ni el cloruro de alquilo ni el alcohol están cargados, el cambio en la polaridad del disolvente no tiene ningún efecto sobre la constante de equilibrio de la reacción. Esto se ilustra dramáticamente observando la velocidad de la reacción en disolventes compuestos de etanol y agua en cantidades variables:
| % agua | 10 | 20 | 30 | 40 | 50 | 60 |
|---|---|---|---|---|---|---|
|
|
1.7 | 9.1 | 40.3 | 126 | 367 | 1294 |