
3.2: Ácidos y bases de Brønsted y Lewis
- Page ID
- 72093

Tres teorías de ácidos y bases
Existen tres clasificaciones principales de sustancias conocidas como ácidos o bases. La definición de Arrhenius establece que un ácido produce H + en solución y una base produce OH -. Esta teoría fue desarrollada por Svante Arrhenius en 1883. Posteriormente, se propusieron dos teorías más sofisticadas y generales. Estas son las definiciones de Brønsted-Lowry y Lewis de ácidos y bases. La relación entre estas teorías se ilustra en la siguiente figura.
|
Ilustración de la jerarquía de las teorías ácido-base. Los ácidos y bases de Arrhenius son una subclase de ácidos y bases de Brønsted, que son a su vez una subclase de ácidos y bases de Lewis. |
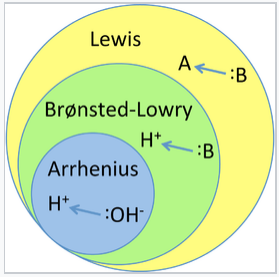
La teoría de Arrhenius, que es la descripción más simple y menos general de ácidos y bases, incluye ácidos como HClO 4 y bases como NaOH o Mg (OH) 2. Esta teoría describe con éxito cómo los ácidos y las bases reaccionan entre sí para hacer agua y sales. Sin embargo, no explica por qué algunas sustancias que no contienen iones hidróxido, por ejemplo F - y NO 2 -, pueden hacer soluciones básicas en agua. La definición de Brønsted-Lowry de ácidos y bases aborda este problema. En esta teoría un ácido es una sustancia que puede liberar un protón (como en la teoría de Arrhenius) y una base es una sustancia que puede aceptar un protón. Una sal básica como Na + F - genera iones OH - en el agua tomando protones del agua misma (para hacer HF):
\[\ce{F^{-}_{(aq)} + H2O_{(l)} <=> HF_{(aq)} + OH^{-}}\]
Cuando un ácido de Brønsted se disocia, aumenta la concentración de iones hidrógeno en la solución, [H +]; a la inversa, las bases de Brønsted se disocian tomando un protón del disolvente (agua) para generar [OH -].
Disociación ácida:\(\ce{HA_{(aq)} <=> A^{-}_{(aq)} + H+_{(aq)}}\)
\[K_{a}= \frac{[A^{-}][H^{+}]}{[HA]}\]
Disociación de bases:\(\ce{B_{(aq)}+ H2O_{(l)} <=> HB+_{(aq)} + OH^{-}+{(aq)}}\)
\[K_{b}= \frac{[HB^{+}][OH^{-}]}{[B]}\]
Ácidos y bases conjugadas
Una consecuencia importante de estos equilibrios es que cada ácido (HA) tiene una base conjugada (A-), y viceversa. En el equilibrio de disociación de bases por encima del conjugado ácido de la base B es HB +.
Para un ácido o base dado, estos equilibrios están unidos por el equilibrio de disociación de agua:
\[\ce{H2O_{(l)} <=> H^{+}_{(aq)} + OH^{-}_{(aq)}}\]
\[K_{w} = [H^{+}][OH^{-}]\]
para lo cual la constante de equilibrio K w es 1.00 x 10 -14 a 25°C.Se puede demostrar fácilmente que el producto de las constantes de disociación ácida y base K a y K b es K w.
Ácidos y bases fuertes y débiles
Se dice que los ácidos y bases que se disocian completamente son fuertes:
\[\ce{HClO4_{(aq)} -> H^{+}_{(aq)} + ClO4^{-}_{(aq)}}\]
\[\ce{HBr_{(aq)} -> H^{+}_{(aq)} + Br^{-}_{(aq)}}\]
\[\ce{CH3O^{-}_{(aq)} + H2O_{(l)} -> CH3OH_{(aq)} + OH^{-}_{(aq)}}\]
\[\ce{NH2^{-}_{(aq)} + H2O_{(l)} -> NH3_{(aq)} + OH^{-}_{(aq)}}\]
Aquí la flecha diestra (→) implica que la reacción va hasta su finalización. Es decir, una solución 1.0 M de HClO 4 en agua en realidad contiene 1.0 M H + (aq) y 1.0 M ClO 4 - (aq), y muy poca HClO 4 no disociada.
Por el contrario, los ácidos débiles como el ácido acético (CH 3 COOH) y las bases débiles como el amoníaco (NH 3) se disocian solo ligeramente en agua -típicamente un poco por ciento, dependiendo de su concentración y los valores de K a y K b - y existen principalmente como las moléculas no disociadas.
|
Los comprimidos antiácidos contienen típicamente sales de calcio del ion bicarbonato (HCO 3 -), una base débil. Su ácido conjugado, el ácido carbónico (H 2 CO 3) es un ácido débil. El equilibrio ácido-base entre el ácido carbónico y el bicarbonato es importante para mantener el pH de la sangre. |

Ejemplo
El amoníaco doméstico es una solución de NH 3 en agua que oscila entre aproximadamente 5-10% en peso. Calculemos el porcentaje de ionización y el pH de la solución.
Solución
Para una solución que sea 8% de amoníaco en peso, asumiendo que la densidad es aproximadamente la misma que la del agua líquida, la concentración analítica de amoníaco es (80 g/L)/(17 g/mol) = 4.7 M.
La otra cosa que necesitamos saber para resolver este problema es la constante de disociación base,\(K_b\).
\(\ce{NH3 + H2O <=> NH4^{+} + OH^{-}} \; K_b = 1.8 \times 10^{-5}\)
Podemos resolver este problema de manera rigurosa invocando tanto el balance de carga ([H +] + [NH 4 +] = [OH -]) como el balance de masa (4.7 M = [NH 3] + [NH 4 +]) y usando\(K_W\) = [H +] [OH -]. Pero debido a que el álgebra se complica con ese método, lo que lleva a una ecuación cúbica que es difícil de resolver, invocaremos dos supuestos simplificadores:
\([NH_{4}^{+}] \approx [OH^{-}] \gg [H^{+}]\)(que es una suposición razonable para una solución básica)
y
\([NH_{3}] \gg [NH_{4}^{+}]\)(también razonable si el porcentaje de ionización es pequeño)
Ahora podemos escribir:
\([NH_{4}^{+}][OH^{-}] \approx [OH^{-}]^{2} = (4.7M)(K_{b})= 8.4 \times 10^{-5}\)
\([OH^{-}]= 9.2 \times 10^{-3} M \; (\approx [NH_{4}^{+}]), \: [H^{+}] = \frac{K_{w}}{[OH^{-}]} = 1.1 \times 10^{-12}M, \: \textbf{ pH = 11.97}\)
El porcentaje de ionización es:
\(100\% \times 9.2 \times 10^{-3}M / 4.7M = \textbf{0.19%}\)
Este ejemplo ilustra que es técnicamente incorrecto etiquetar una botella de amoníaco acuoso como “hidróxido de amonio”, ya que solo aproximadamente 2/10 del uno por ciento de la base débil existe en esa forma.
Ácidos y bases conjugadas
Un error común es que los ácidos fuertes tienen bases conjugadas débiles y que los ácidos débiles tienen bases conjugadas fuertes. Es fácil ver que esto es incorrecto al recordar que K a K b = K w. Nuestra definición de un ácido o base fuerte es que K >> 1, es decir, que la sustancia se disocia completamente. Nuestra definición de un ácido o base débil es 1 > K > K w. De ello se deduce que si K a >> 1 (fuerte) entonces K b no puede ser > K w (débil).
De hecho, los ácidos fuertes como el HCl se disocian para producir iones espectadores como Cl - como bases conjugadas, mientras que los ácidos débiles producen bases conjugadas débiles. Esto se ilustra a continuación para el ácido acético y su base conjugada, el anión acetato. El ácido acético es un ácido débil (K a = 1.8 x 10 -5) y el acetato es una base débil (\(K_{b} = \frac{K_{w}}{K_{a}} = 5.6 \times 10^{10}\))

La fuerza de un conjugado ácido/base varía inversamente con la fuerza o debilidad de su ácido o base parental. Cualquier ácido o base es técnicamente un ácido conjugado o una base conjugada también; estos términos se usan simplemente para identificar especies en solución (es decir, el ácido acético es el ácido conjugado del anión acetato, una base, mientras que el acetato es la base conjugada del ácido acético, un ácido).
Los oxiácidos neutros (H 2 SO 4, H 3 PO 4, HNO 3, HClO 2, etc.) pueden clasificarse como fuertes o débiles siguiendo una regla simple señalada por primera vez por Linus Pauling. Si el número de átomos de oxígeno excede el número de átomos de hidrógeno en dos o más, entonces el ácido es fuerte; de lo contrario es débil. Por ejemplo HClO 4 y HClO 3, donde la diferencia es 3 y 2, respectivamente, son ambos ácidos fuertes. HNO 2 y HClO 2 son débiles porque la diferencia es 1 en ambos casos. Para los ácidos débiles, la fuerza relativa depende de esta diferencia (es decir, HClO 2 es un ácido débil más fuerte que el HOCl) y de la electronegatividad del átomo central (HOCl es más fuerte que HOI).
Los ácidos que pueden donar más de un protón se denominan ácidos polipróticos. Por ejemplo, el ácido sulfúrico, H 2 SO 4, es un ácido fuerte que tiene una base conjugada que en realidad resulta ser un ácido débil en sí mismo. Esto significa que cada mol de H 2 SO 4 en solución acuosa dona más de 1 mol de protones. El ácido carbónico (H 2 CO 3) y el ácido fosfórico (H 3 PO 4) son ácidos polipróticos débiles. Típicamente, los pK a secuenciales de ácido poliprótico están separados por aproximadamente 5 unidades de pH, debido a que progresivamente se vuelve más difícil eliminar protones a medida que el ion se carga más negativamente. Por ejemplo, los tres pK a de ácido fosfórico son 2.15, 7.20 y 12.35.
Compuestos anfóteros
Algunas sustancias pueden actuar ya sea como ácido y como base. Un ejemplo es el agua. Las moléculas H 2 O pueden donar un ión hidrógeno o aceptar uno. Esta propiedad hace que el agua sea un solvente anfótera. En la situación en la que un ácido se disocia en solución, el agua está actuando como base. Por el contrario, el agua actúa como un ácido cuando las bases se disocian. El ácido más fuerte que podemos hacer en H 2 O es H + (aq), y la base más fuerte que podemos hacer en H 2 O es OH - (aq).
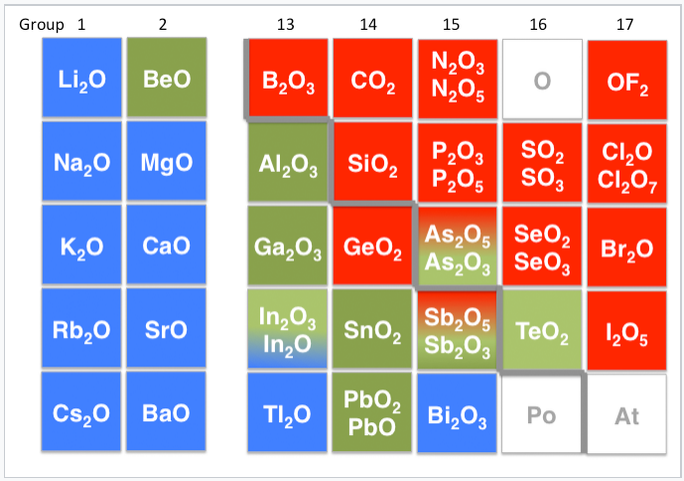
Otros ejemplos de compuestos anfóteros son óxidos e hidróxidos de elementos que se encuentran en el límite entre los elementos metálicos y no metálicos en la tabla periódica. Por ejemplo, el hidróxido de aluminio (Al (OH) 3) es insoluble a pH neutro, pero puede aceptar protones en ácido para hacer [Al (H 2 O) 6] 3+ o aceptar un ion OH - en base para formar iones Al (OH) 4 -. En consecuencia, el óxido de aluminio es soluble en ácido y en base, pero no en agua neutra. Otros ejemplos de óxidos anfóteros son BeO, ZnO, Ga 2 O 3, Sb 2 O 3 y PbO. Al aumentar el estado de oxidación de un metal se incrementa la acidez de su óxido al retirar la densidad electrónica de los átomos de oxígeno. Así, Sb 2 O 5 es ácido, pero Sb 2 O 3 es anfótero.
|
Tabla periódica que muestra los óxidos básicos (azul), anfótero (verde) y ácidos (rojos). El límite metal-no metálico está indicado por la línea gris de la escalera. |
Nivelación de solventes
La nivelación del solvente es un efecto que ocurre cuando se coloca un ácido fuerte en un solvente como (pero no limitado a) H 2 O. Debido a que los ácidos fuertes donan sus protones al solvente, el ácido más fuerte posible que puede existir es el ácido conjugado del solvente. En solución acuosa, este es H 3 O +. Esto significa que la fuerza de ácidos como HCl y HBr no se puede diferenciar en agua ya que ambos están disociados 100% a H 3 O +. En el contexto de nuestra discusión sobre las bases conjugadas anteriormente, diríamos que tanto Cl - como Br - son iones espectadores en el agua: ninguno es una base lo suficientemente fuerte como para aceptar un protón de H 3 O +. Para diferenciar las acideces de ácidos fuertes como HClO 4 y HCl, o las basicidades de bases fuertes como CH 3 O - y NH 2 -, normalmente debemos trabajar en disolventes no acuosos, como se explica a continuación.
Soluciones no acuosas
La teoría de Brønsted abarca cualquier tipo de disolvente que pueda donar y aceptar iones H +, no solo soluciones acuosas. La fuerza de un ácido o una base varía dependiendo del disolvente. La química ácido-base no acuosa sigue reglas similares a las desarrolladas para ácidos y bases en agua. Por ejemplo, en amoníaco líquido, el disolvente se autodisocia en la reacción:
\[\ce{2NH3_{(l)} <=> NH4^{+} + NH2^{-}}\]
Este equilibrio es análogo a la autodisociación del agua, pero tiene una constante de equilibrio menor (K ≈ 10 -30). Se deduce por analogía al agua que NH 4 + es el ácido más fuerte y NH 2 - es la base más fuerte que puede existir en el amoníaco líquido. Debido a que el amoníaco es un solvente básico, potencia la acidez y suprime la basicidad de las sustancias disueltas en él. Por ejemplo, el ion amonio (NH 4 +) es un ácido débil en el agua (K a = 6 x 10 -10), pero es un ácido fuerte en amoníaco. De igual manera, el ácido acético es débil en el agua pero fuerte en amoníaco. De hecho, la nivelación de solventes hace que HCl, CH 3 COOH y NH 4 Cl sean ácidos fuertes en amoníaco, donde tienen una fuerza ácida equivalente.
Los ácidos fuertes que se nivelan en agua tienen diferentes resistencias ácidas en disolventes ácidos como HF o ácido acético anhidro. Por ejemplo, la disociación ácida de HX en ácido acético (CH 3 COOH) implica protonar el disolvente para hacer su conjugado ácido (CH 3 COOH 2 +) y el anión X -. Debido a que CH 3 COOH 2 + es un ácido más fuerte que H 3 O +, el anión X - (que es un espectador en el agua) puede convertirse en una base débil en CH 3 COOH:
\[\ce{HX + CH3COOH <=> CH3COOH2^{+} + X^{-}}\]
De ello se deduce que los disolventes ácidos magnifican las basicidades de Brønsted de sustancias que no pueden aceptar protones en el agua. Por el contrario, los solventes básicos magnifican la acidez de sustancias que no pueden donar un protón al OH -.
La acidez y basicidad de los disolventes no acuosos es difícil de cuantificar con precisión, pero una buena medida relativa es la función de acidez de Hammett, H o. H o se define análogamente al pH de acuerdo con la ecuación de Henderson-Hasselbach:
\[H_{o} = pK_{a} + \log(\frac{[base]}{[conjugate \: acid]})\]
Para disolventes no acuosos, o para compuestos ácidos o básicos disueltos en disolventes que no se disocian por sí mismos, el H o es una medida aproximada del pH del disolvente o compuesto en cuestión. El HF anhidro y H 2 SO 4 tienen valores de H o de aproximadamente -10 y -12 respectivamente.
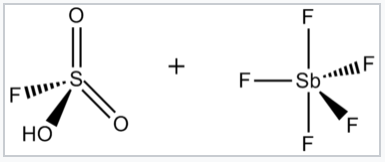
Superácidos y superbases. El término superácido fue acuñado originalmente por James Bryant Conant en 1927 para describir ácidos que eran más fuertes que los ácidos minerales convencionales. [1] George A. Olah preparó el llamado ácido mágico, llamado así por su capacidad para atacar hidrocarburos, mezclando pentafluoruro de antimonio (SbF 5) y ácido fluorosulfónico (FSO 3 H). El nombre fue acuñado después de que se colocara una vela en una muestra de ácido mágico. La vela se disolvió, mostrando la capacidad del ácido para protonar hidrocarburos, que bajo condiciones ácidas acuosas no pueden protonarse.
|
El ácido mágico se elabora mezclando FSO 3 H y SbF 5. Su reacción genera el catión H 2 SO 3 F +, que puede protonar hidrocarburos. |
A 140 °C, FSO 3 H—SbF 5 convierte el metano en el carbocatión de butilo terciario, una reacción que comienza con la protonación del metano: [2]
\[\ce{CH4 + H^{+} -> CH5^{+}}\]
\[\ce{CH5^{+} -> CH3^{+} + H2}\]
\[\ce{CH3^{+} + 3CH4 -> (CH3)3C^{+} + 3H2}\]
El ácido fluoroantimónico, HSbF 6, puede producir soluciones con H 0 hasta —28. [3] El ácido fluoroantimónico se elabora combinando HF y SbF 5. En este sistema, HF libera su protón (H +) concomitante con la unión de F − por pentafluoruro de antimonio, que (como se describe a continuación) es un ácido de Lewis. El anión resultante (SbF 6 -) es tanto un nucleófilo débil como una base extraordinariamente débil.
Los superácidos son útiles en reacciones como la isomerización de alcanos. Industrialmente, las zeolitas anhidras intercambiadas con ácido, que son catalizadores superácidos, se utilizan a escala masiva para isomerizar hidrocarburos en el procesamiento de petróleo crudo a gasolina. Superbases como dietilamida de litio (LiNet 2), compuestos de alquillitio (RLi) y reactivos de Grignard (rMgX) útiles en una amplia gama de reacciones orgánicas. LiNet 2 desprotonata los enlaces C-H para generar carbaniones reactivos. RLi y RMGx son potentes nucleófilos.
El uso de superbases en medios no acuosos nos permite clasificar las acidedades (y medir los pKa) de diferentes clases de moléculas. Esta clasificación es particularmente importante para comprender las reacciones de las moléculas orgánicas. Tenga en cuenta que el orden de acidedades para los hidrocarburos es alquinos >> alquenos, aromáticos >> alcanos. Este ordenamiento tiene que ver con la hibridación del átomo de carbono que forma el carbanión. El par solitario cargado negativamente del carbanión se estabiliza en orbitales que tienen alto carácter s (e.g., sp vs. sp 2 o sp 3). Esto se debe a que los orbitales tienen densidad de probabilidad finita en el núcleo y “sienten” la carga nuclear positiva (estabilizando así la carga extra negativa sobre el carbono) más que p orbitales. Los efectos de resonancia también estabilizan los carbaniones. Así, el ciclopentadieno es más ácido que incluso un alquino debido a que la carga negativa se deslocaliza sobre todo el anillo (aromático) C 5 H 5 - cuando el C 5 H 6 está desprotonado.
| nombre | fórmula | fórmula estructural | pKa |
|---|---|---|---|
| Metano | CH 4 |  |
56 |
| Propeno | C 3 H 6 | 44 | |
| Benceno | C 6 H 6 | 43 | |
| Acetileno | C 2 H 2 |  |
25 |
| Ciclopentadieno | C 5 H 6 |  |
18 |
Cuadro 1. Acidedades ácidas de carbono en pKa en DMSO [4].
Equilibrios ácido-base en sales fundidas
Cuando una sal sólida se funde, forma una solución de los cationes y aniones. Por ejemplo, el KOH se funde a temperaturas superiores a 400 °C y se disocia en iones K + y OH - que pueden actuar como disolvente para reacciones químicas. Debido a la autodisociación del disolvente OH -, el agua siempre está presente en un flujo de KOH fundido, de acuerdo con el equilibrio ácido-base:
\[\ce{2 OH^{-} <=> H2O + O^{2-}}\]
De ello se deduce que en este disolvente muy básico, el agua (el ácido conjugado del disolvente) es el ácido más fuerte que puede existir. La base conjugada del disolvente, O 2-, es la base más fuerte. Este equilibrio de autodisociación permite que la acidez de un flujo se ajuste fácilmente a través de la adición o ebullición del agua. Un fundente “húmedo” es más ácido y puede disolver óxidos metálicos que contienen el anión O 2- básico. Por el contrario, un flujo “seco” es más básico y provocará que precipiten los óxidos. Así, los fundentes de hidróxido fundido pueden ser utilizados en la síntesis de cristales de óxido, como el superconductor de perovskita (K 1- X Ba X BiO 3). [5]. Las mezclas eutécticas de NaOH y KOH tienen un punto de fusión relativamente bajo (≈ 200 °C) y pueden usarse como solventes para cristalizar una variedad de óxidos básicos.
Ácidos y bases de Lewis
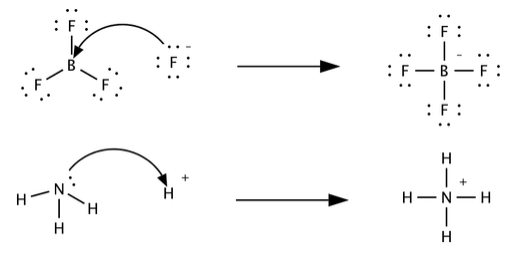
La clasificación de Lewis de ácidos y bases es más amplia que la definición de Brønsted-Lowry, y abarca muchas más sustancias. Mientras que las clasificaciones de Brønsted-Lowry y Arrhenius se basan en la transferencia de protones, la acidez y basicidad de Lewis se basan en la compartición de un par de electrones. Los ácidos de Lewis pueden aceptar un par de electrones, mientras que las bases de Lewis pueden donar un par de electrones. Esta definición abarca la definición de Brønsted-Lowry, en que H + es un aceptor de pares de electrones (cuando interactúa con una base), y una base es un donante de pares de electrones en su interacción con H +. Esto se ilustra a continuación para la protonación del amoníaco.
Trifluoruro de boro, BF 3 actúa como un ácido de Lewis cuando se combina con un ion básico o molécula que puede donar un par de electrones. Dicha reacción se muestra a continuación.
\ [\ ce {BF3 + F^ {-} <=> BF4^ {-}
Aquí, el ácido es BF 3 y la base es F -. Esta reacción ácido-base permite que el boro (que es deficiente en electrones en BF 3) complete su octeto. De manera similar, AlCl 3 es un ácido de Lewis que puede reaccionar con Cl - (una base de Lewis) para hacer la “sal” de Lewis AlCl 4 -. Tenga en cuenta que en el agua Cl - es un ion espectador (una base más débil que el disolvente) en las reacciones ácido-base de Brønsted.
Ejemplos adicionales de reacciones ácido-base de Lewis. En cada uno, tratar de identificar el ácido, la base y la sal, con base en el concepto de que la base es la molécula o ion que dona un par de electrones. En los casos en los que no esté seguro, puede ayudar dibujar las estructuras VSEPR de las moléculas:
\(\ce{I2 + I^{-} <=> I3^{-}}\)
\(\ce{AuCl3 + Cl^{-} <=> [AuCl4]^{-}}\)
\(\ce{Fe^{3+} + 6H2O <=> [Fe(H2O)6]^{3+}}\)
\(\ce{TiF4 + 2F^{-} <=> [TiF6]^{2-}}\)
\(\ce{SF4 + SbF5 <=>SSbF9}\)
En otras reacciones ácido-base de Lewis, tanto el ácido como la base son moléculas y el producto se denomina aducto.
\(\ce{(CH3)3B + N(CH3)3 -> (CH3)3B-N(CH3)3}\)
\(\ce{I2 + S(CH3)2 -> I2-S(CH3)2}\)
\(\ce{C5N5N + Cu(HFacac)2 -> C5N5N-Cu(HFacac)2}\)
La acidez de Lewis es la base de la química de coordinación, tema que discutiremos con más detalle en el Capítulo 5. Esto se debe a que la química de coordinación involucra iones metálicos que son ácidos de Lewis, que se unen a ligandos que son bases de Lewis.
Determinación de la fuerza de los ácidos de Lewis de iones metálicos
Hay tres factores determinantes en la fuerza ácida de Lewis de un ión metálico:
1. Cuanto mayor sea la carga positiva sobre el metal, más ácida es. Por ejemplo, Al 3 + y Fe 3 + son buenos ácidos de Lewis y sus sales hacen soluciones ácidas en agua, pero K + y Na + no lo son.
2. Cuanto menor es el radio atómico del ión metálico, más ácido es. Bajando por la tabla periódica, la acidez de Lewis de los iones metálicos disminuye (e.g., Al 3 + > Ga 3 + > In 3 +) debido a que el radio iónico aumenta.
3. Para los iones de metales de transición, los metales más electronegativos tienden a hacer ácidos de Lewis más fuertes. La electronegatividad tiene máximos en W y Au en la serie 5d, por lo que los iones metálicos cercanos en esa parte de la tabla periódica son buenos ácidos de Lewis.
Las moléculas con cinco geometrías de coordenadas (p. ej., PCl 5, AsF 5, SbF 5) son típicamente ácidos de Lewis fuertes, ya que al aceptar otro par de electrones de una base, forman una molécula octaédrica o anión. Ninguna de las geometrías comunes de cinco coordenadas (bipiramidal trigonal o piramidal cuadrado) es eficiente en términos de empaque. La reacción ácido-base de Lewis forma un enlace adicional con una penalización energética relativamente pequeña de estirar los enlaces existentes:
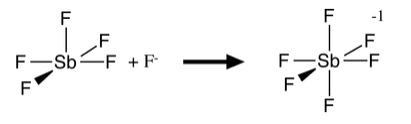
Debido a que F - es una buena base de Lewis, y también un anión pequeño, puede formar aniones octaédricos estables tanto con elementos del grupo principal como con metales de transición. Por esta razón, TiO 2 y SiO 2 se disuelven en HF (pero no son reactivos con HCl acuoso y otros ácidos fuertes):
\[\ce{TiO2 + 4HF + 2F^{-} <=> TiF6^{2-} + 2H2O}\]
\[\ce{SiO2 + 4HF + 2F^{-} <=> SiF6^{2-} + 2H2O}\]
Las bases de Lewis estabilizan estados de alta oxidación.
Un ejemplo interesante del uso de la química ácido-base de Lewis para impulsar reacciones es la síntesis química del gas flúor, que fue ideada por Karl O. Christe en 1986. [6] Christe en su momento estaba organizando un simposio para conmemorar el centenario del aislamiento del flúor elemental por Henri Moissan, lo que Moissan hizo en 1886 electrolizando una solución de HF anhidro. 100 años después, aún no hubo síntesis directa (no electroquímica) de F 2. El esquema de reacción de Christe siguió dos pasos. El primero fue la síntesis conocida de K 2 MnF 6 a partir de KMnO 4:
\[\ce{4MnO4^{-}_{(aq)} + 10H2O_{(l)} + 24F^{-}_{(aq)} -> 4MnF6^{2-}_{(aq)} = 3O2_{(g)} + 20OH^{-}_{(aq)}}\]
\[\ce{2K^{+}_{(aq)} + MnF6^{2-}_{(aq)} -> K2MnF6_{(s)}}\]
El segundo paso consistió en hacer reaccionar K 2 MnF 6 con el potente ácido de Lewis SbF 5, para hacer metaestable MnF 4, que se descompone espontáneamente en MnF 3 y gas flúor:
\[\ce{K2MnF6_{(s)} + 2SbF5_{(l)} -> 2KSbF6_{(s)} + MnF4_{(s)}}\]
\[\ce{MnF4_{(s)} -> MnF3_{(s)} + \frac{1}{2}F2_{(g)}}\]
Esta reacción nos enseña algo interesante e importante sobre la conexión entre la química ácido-base y la química redox. Los ácidos tienden a estabilizar estados de baja oxidación, y las bases estabilizan estados de oxidación alta (Lo veremos de nuevo pronto en el Capítulo 4, en el contexto de los diagramas de Pourbaix). El Mn es estable en el estado de oxidación +4 en K 2 MnF 6, donde está rodeado por seis aniones F básicos. Sin embargo, el fluoruro neutro más estable de Mn es MnF 3, y MnF 4 (transitoriamente formado a partir de K 2 MnF 6) se descompone espontáneamente para generar flúor.
El óxido es una mejor base que el fluoruro. Curiosamente, Mn puede perder todos sus electrones de valencia para formar Mn 7 + en el ion permanganato, MnO 4 -. Aquí el estado de oxidación 7+ se estabiliza electrostáticamente por coordinación a cuatro iones O 2-, y por la carga general -1 en el anión MnO 4 -. Debido a su 2- carga, O 2- es una base más fuerte y un mejor ion para estabilizar estados de alta oxidación que F -. Esta es una tendencia general entre los metales de transición: el estado de oxidación más alto generalmente se alcanza en el óxido, no en el fluoruro, a pesar de que F es un elemento más electronegativo que O. Por ejemplo, Cr 6 + es estable en el CrO 4 2 - y Cr 2 O 7 2 - aniones, pero no en ningún fluoruro neutro o fluoroanión. El estado de oxidación +8 ocurre en RuO 4 y OsO 4, pero no en ningún fluoruro de Ru u Os.