1.9: Teoría de Bandas en Sólidos
- Page ID
- 74673

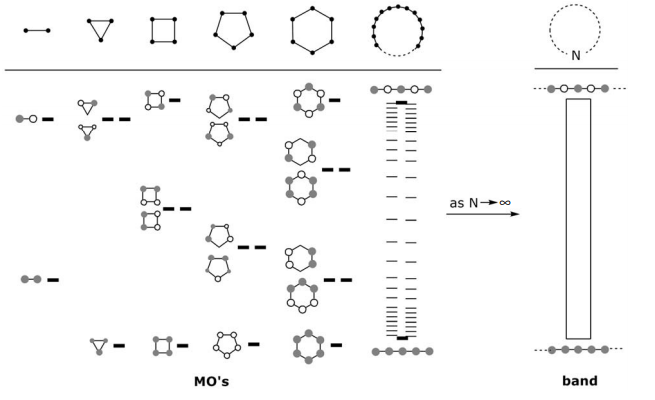
El método LCAO para sistemas cíclicos proporciona un punto de partida conveniente para el desarrollo de la estructura electrónica de sólidos.
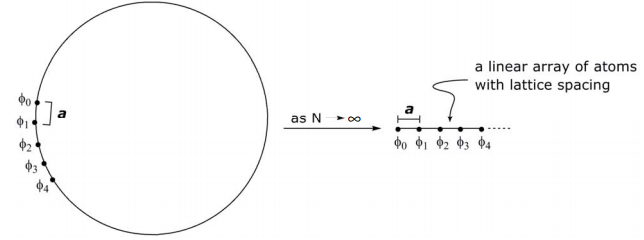
A N muy grande, a medida que la circunferencia del círculo se acerca ∞, el problema cíclico converge a uno lineal,
Cualitativamente, desde una perspectiva de nivel de energía MO,
Más cuantitativamente, al pasar de sistemas cíclicos a lineales, en lugar de describir posiciones orbitales (átomos) angularmente, la posición de un átomo se describe por m a, donde m es el número del átomo en la matriz y a es la distancia entre átomos. Así, la θ de la derivación N-cíclica se convierte en m a,
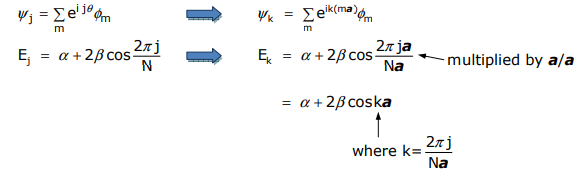
Unas palabras sobre\(k\). Es:
- una medida del número de nodos
- un índice de función de onda y, en consecuencia, simetría de función de onda
- un “número cuántico” para un ψ k dado
- una medida de longitud, relacionada con la longitud de onda λ —1
- de la relación de DeBroglie, λ = (h/p), por lo tanto k es también un vector de onda que mide el impulso
Volviendo a la discusión anterior, tenga en cuenta que k paramétricamente depende de a. Dado que a es un parámetro de celosía de la celda unitaria, hay tantas k como celdas unitarias en el cristal. En el caso lineal, la celda unitaria es la distancia entre los átomos adyacentes: hay n átomos n celdas unitarias o en otros términos — hay tantas k como átomos en la cadena 1-D.
Determinemos los valores energéticos de los límites, k = 0 y k = (π/ a):
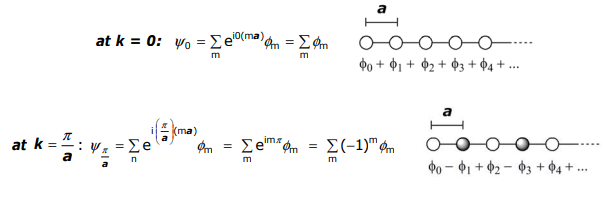
Las energías para estas estructuras de banda en los límites de k son:
\[ E_{0} = \alpha + 2\beta \cos(0)a = \alpha + 2\beta \]
\[ E_{ \dfrac{ \pi }{a}} = \alpha + 2\beta \cos( \dfrac{ \pi }{a} )a = \alpha - 2\beta \]
Nótese que k está cuantificado; por lo que hay un número finito de valores entre α+2β y α—2β pero para un número muy grande (~10 23 átomos) entre los límites de k, así, la energía es una función continua y que varía suavemente entre estos límites.
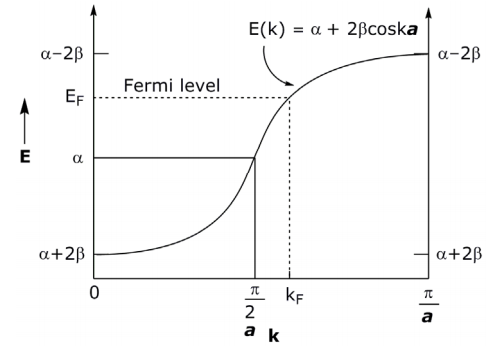
El rango - π/ a ≤ k ≤ π/ a o |k| ≤ π/ a es único porque la función se repite a a a a fuera de estos límites. Este rango único de valores k se llama la zona Brillouin. La primera zona de Brillouin se representa arriba de 0 a π/ a (reflexión simétrica de − π/ a a a 0).
Con un número dado de e — s en el sólido, los niveles se llenarán a una cierta energía llamada nivel Fermi, que corresponde a un cierto valor de k (= k F). En el ejemplo k anterior, hay más electrones que orbitales, así que k F > k/2 a. Si cada orbital atómico contribuyó 1e — al sistema, entonces E F ocurriría para k F = π/2 a.
La simetría de los orbitales atómicos individuales determina mucho sobre la estructura de bandas. Considera que los orbitales p se superponen en una matriz lineal (frente a los orbitales 1s del tratamiento anterior). Analizando formas limitantes:
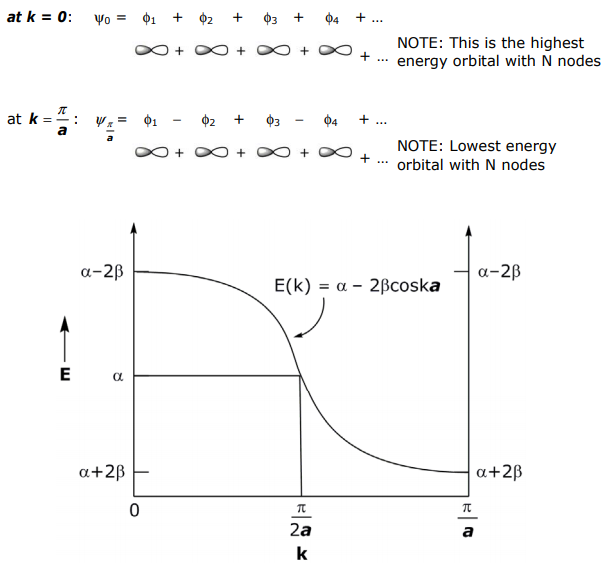
La banda de energía es opuesta a la del LCAO orbital sσ porque el (+) LCAO para un orbital pσ es antiadherentes.
Así, las moléculas se relacionan fácilmente con los sólidos a través de la teoría de Hückel. No es sorprendente que exista un lenguaje de química que describe la estructura electrónica de las moléculas que se relaciona con el lenguaje de la física que describe la estructura electrónica de los sólidos. A continuación se presentan algunos de los términos que los químicos y físicos utilizan para describir fenómenos similares en moléculas y sólidos:
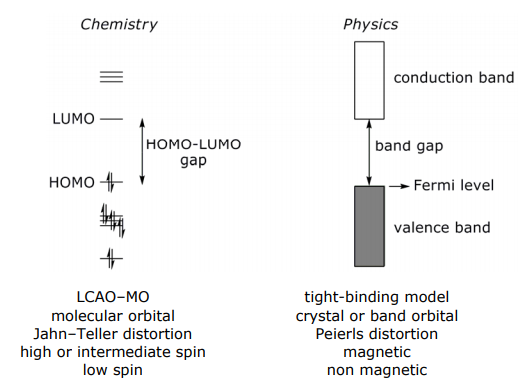
Ancho de Banda o Dispersión
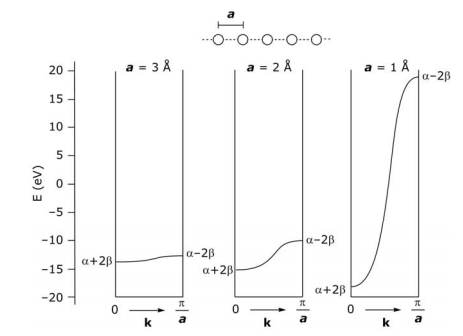
¿Qué determina el ancho o dispersión de una banda? En cuanto a la brecha HOMO-LUMO en una molécula, el solapamiento de orbitales vecinos determina la dispersión de energía de una banda; cuanto mayor sea el solapamiento, mayor será la dispersión. Observe cómo la dispersión de banda de una cadena lineal de átomos de H varía a medida que los orbitales 1s de los átomos de H están separados 1, 2, 3 Å (E de un átomo de H aislado es —13.6 eV):
Densidad de Estados
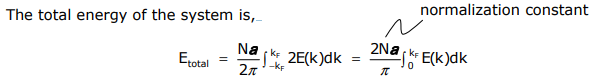
o en otras palabras, es el área bajo la curva a kF. Otra cantidad útil es el número de orbitales entre E (k) + dE (k), denominado densidad de estados (DOS). Para un sistema 1-D,
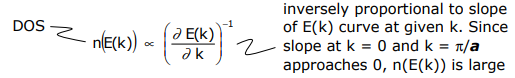
Una gráfica de las ecuaciones anteriores es,
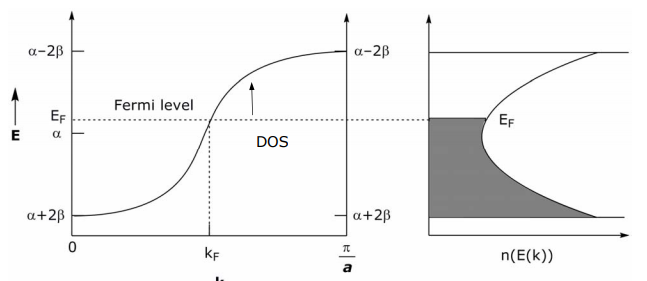
En el diagrama DOS anterior, ninguna brecha de energía separa las bandas llenas y vacías, es decir, hay una densidad continua de estados — esta propiedad es característica de un metal. Si está presente un hueco de energía entre orbitales llenos y vacíos y puede superarse térmicamente, entonces es semiconductor; un hueco de energía que no se puede superar es un aislante.
Un ejemplo 1-D
Podría decirse que el sistema 1-D más conocido en química inorgánica es K 2 Pt (CN) 4 y su compuesto parcialmente oxidado (por ejemplo, K 2 Pt (CN) 4 Br 0.3).
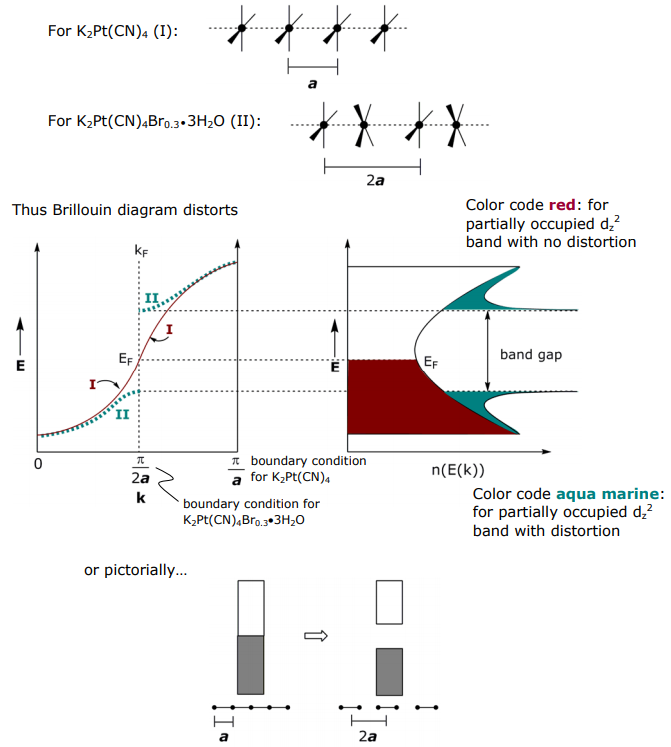
Platinocianido normal, K 2 Pt (CN) 4:
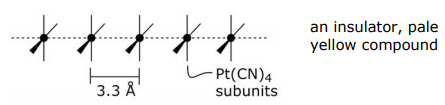
Platinocianido parcialmente oxidado, K 2 Pt (CN) 4 Br 0.3 •3H 2 O:

Nota: d (Pt-Pt) = 2.78 Å en metal Pt
Para explicar estas dispares propiedades de los compuestos 1-D, considere la subunidad molecular Pt (CN) 4 2-:
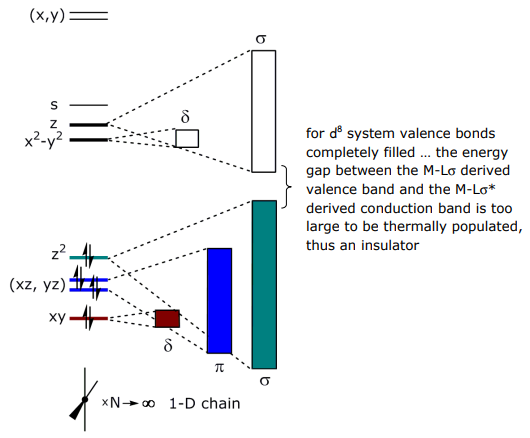
La dispersión de las bandas se debe a las diferentes superposiciones de los orbitales dσ, dπ y dδ.
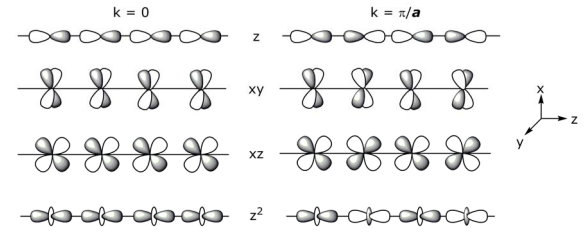
La estructura de bandas (o primeras zonas Brillouin) derivada de las MO fronterizas es:
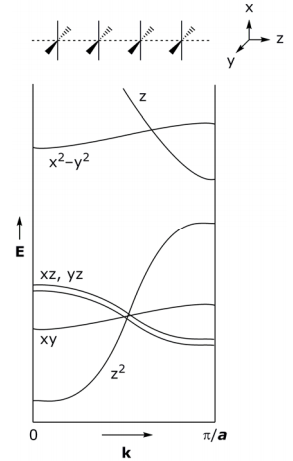
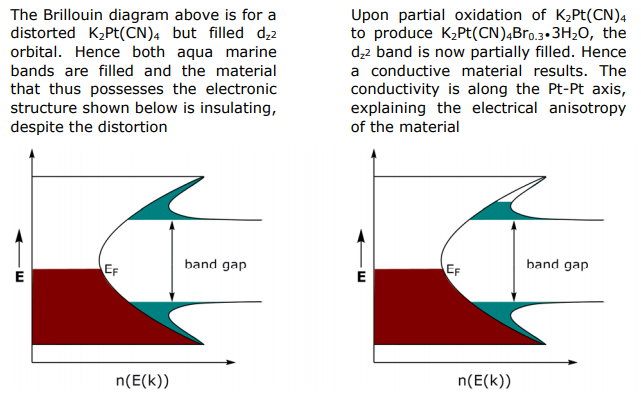
Para el sistema parcialmente oxidado, el enlace σ derivado de dz 2 debe estar parcialmente lleno y por lo tanto metálico, pero no lo es, parcialmente oxidado K 2 Pt (CN) 4 Br x es un semiconductor. Para explicar esta anomalía, considere cómo la estructura de la banda se perturbe ante la oxidación parcial:
Aislando en la banda d z 2 en K 2 Pt (CN) 4, los átomos de Pt están espaciados uniformemente con la dimensión reticular a (I). Tras la oxidación, la cadena de Pt puede distorsionarse para dar una dimensión reticular 2 a (II). En el caso del K 2 Pt (CN) 4 Br 0.3 •3H 2 O la distorsión es una rotación de las subunidades Pt y la formación de dímeros dentro de la cadena, por lo que la dimensión de la celda unitaria está anclada a cada otro átomo de Pt.