
13.4.2: Ligandos similares al CO
- Page ID
- 81098
Ahora dejemos el\(\ce{CO}\) ligando, consideremos varios ligandos relacionados y discutamos similitudes y diferencias en comparación con el\(\ce{CO}\) ligando.
El ligando ciano (CN -)
Empecemos con el ligando ciano\(\ce{CN^-}\). Este ligando es isoelectrónico para el\(\ce{CO}\) ligando. El átomo de N tiene un electrón menor que O, pero la carga negativa en el ligando de cianuro compensa eso. Una pregunta que podemos hacer es: ¿Cuál es el final reactivo? La respuesta es: En analogía con el ligando carbonilo el extremo reactivo es el átomo de carbono. Podemos explicar esto por el hecho de que el diagrama MO es similar al de\(\ce{CO}\), solo las diferencias en las energías orbitales atómicas son menores debido a la menor diferencia de electronegatividad entre C y N en comparación con C y O. Por lo tanto, como en\(\ce{CO}\), el HOMO está representado por el par solitario de electrones en la C, lo que hace que C sea el extremo más reactivo. Debido a la menor diferencia de electronegatividad, la diferencia de energía entre el par solitario en C y el par solitario en N es menor, por lo tanto, en contraste con\(\ce{CO}\), el ligando ciano actúa mucho más a menudo como un ligando puente entre dos metales usando sus dos pares solitarios de electrones.
¿Esperaríamos que el ligando ciano sea un\(\sigma\) donante más fuerte o más débil en comparación con el\(\ce{CO}\) ligando? Piénsalo un momento. La respuesta es: Es un\(\sigma\) donante más fuerte por su carga negativa. La carga negativa en el ligando aumenta la repulsión electrostática entre los electrones, y esto aumenta las energías orbitales. Por lo tanto, existe una tendencia más fuerte a donar los electrones. Nuestra siguiente pregunta es: ¿Es el\(\ce{CN^-}\) ligando un\(\pi\) aceptor más fuerte o más débil que\(\ce{CO}\)? La energía de los\(\pi^*\) orbitales es mayor en comparación con\(\ce{CO}\) debido a la carga negativa en el ligando. Debido a eso, los electrones del metal no pueden ser tan fácilmente aceptados por el ligando. Por lo tanto, el ligando ciano es un\(\pi\) aceptor más débil que el ligando carbonilo. Nuestra última pregunta es: ¿Son los complejos ciano más estables con metales en estados de oxidación alta o baja. Debido a argumentos electrostáticos, un anión cianuro interactúa más fuertemente con un catión metálico que con un metal en un estado de oxidación cero o negativo. Por lo tanto, a diferencia\(\ce{CO}\),\(\ce{CN^-}\) no estabiliza los metales en estados de baja oxidación. Prefiere hacer complejos con metal en altos números de oxidación positivos.
El Ligando Nitrosil NO
El ligando nitrosil\(\ce{NO}\) es otro ligando similar a\(\ce{CO}\). Tiene un electrón más que\(\ce{CO}\) debido a que N tiene un electrón más que C. El electrón adicional hace\(\ce{NO}\) una molécula “impar” con un electrón radical. Al igual que en\(\ce{CO}\) y\(\ce{CN^-}\), el elemento más electropositivo es el extremo reactivo. En el caso de\(\ce{NO}\) ello es el átomo de N. El electrón radical es el electrón más reactivo que se puede donar más fácilmente, sin embargo, el par solitario de electrones en N puede ser donado además. En el primer caso, el\(\ce{NO}\) es un donante de 1 electrón, en el segundo es un donante de 3 electrones. ¿Cómo podemos saber si se han donado uno o tres electrones? Cuando solo se dona un electrón, el par solitario de electrones en el nitrógeno es estéricamente activo y conduce a una estructura doblada (Fig. \(\PageIndex{1}\)).
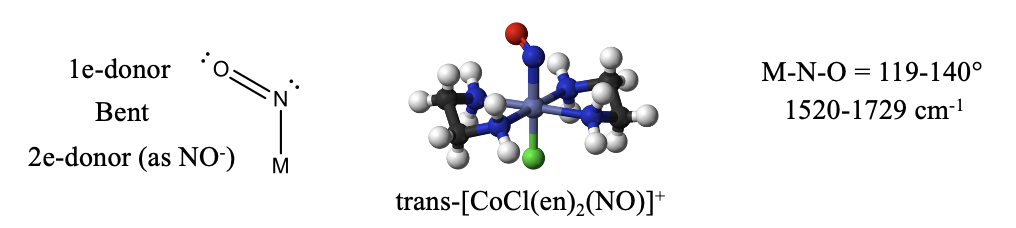
Un ejemplo es el catión trans-bis- (etilendiamina) cloronitrosil cobalto (1+). Generalmente, el ángulo de\(\ce{O-N-M}\) unión en los complejos de nitrosil con\(\ce{NO}\) un 1e-donador puede variar entre 119 y 140°. También podemos identificar un ligando nitrosil doblado en el espectro IR. Los números de onda típicos son 1520-1729 cm -1.
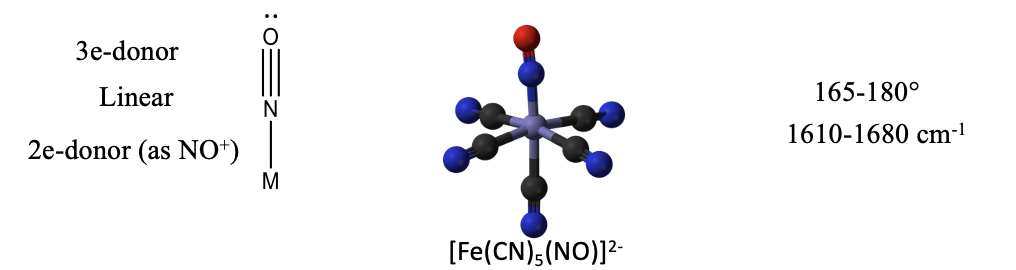
Cuando el ligando nitrosil dona los tres electrones, entonces se une al metal de manera lineal (Fig. \(\PageIndex{2}\)). El par solitario de electrones ya no es estéricamente activo porque está involucrado en la unión. Un ejemplo es el anión nitroprusido\(\ce{[Fe(CN)5(NO)]^2-}\). Se utiliza como medicamento para el tratamiento de la presión arterial alta. Los ángulos de enlace en complejos con\(\ce{NO}\) el donador de 3 electrones a menudo no son exactamente 180°, sino que varían entre 165 y 180°. También se puede identificar en el IR debido a que tiene una banda característica entre 1610-1680 cm -1.
También hay una visión diferente sobre los complejos de nitrosil con estructuras lineales y dobladas. En las estructuras dobladas también podemos considerar al ligando como un\(\ce{NO^-}\) anión que dona dos electrones. El\(\ce{NO^-}\) anión tiene dos pares solitarios de electrones en N. Cuando dona dos electrones entonces un par solitario de electrones estéricamente activos permanece en el átomo de nitrógeno. En estructuras lineales podemos considerar al ligando también como un\(\ce{NO^+}\) catión que dona dos electrones. Un\(\ce{NO^+}\) catión tiene un par solitario de electrones en N, y cuando dona ese par solitario entonces no hay ningún electrón estéricamente activo en el nitrógeno que queda, y así el ligando se une de manera lineal.
También podríamos preguntar: ¿Qué puede ser neutral\(\ce{NO}\) no ser un donante de 2 electrones con el electrón radical dejado en N? La respuesta es: El electrón radical es el electrón de mayor energía, y siempre se usa primero en interacciones con un metal.
Ligandos de fosfina
Las fosfinas son más notables por su notable sintonización electrónica y estérica y su “inocencia”, tienden a evitar participar directamente en reacciones organometálicas, pero tienen la capacidad de modular profundamente las propiedades electrónicas del centro metálico al que están unidas. Además, debido a que la barrera energética para el volteo de las fosfinas es bastante alta, los ligandos “quirales en fósforo” pueden aislarse en forma enantioenriquecida e introducirse en centros metálicos, trayendo asimetría casi tan cerca del metal como puede llegar a los complejos quirales. La RMN de fósforo es una técnica que Just Works (gracias, naturaleza). Las fosfinas blandas coinciden muy bien con los metales de transición blandos de baja valencia. ¡Las fosfinas pobres en electrones son incluso buenos ácidos π!
Al igual que el CO, las fosfinas son ligandos dativos de tipo L que aportan formalmente dos electrones al centro metálico. A diferencia del CO, la mayoría de las fosfinas no son lo suficientemente pequeñas como para formar más de cuatro enlaces a un solo centro metálico (y para R grandes, el número es aún menor). El obstáculo estérico se convierte en un problema cuando cinco o más ligandos PR 3 intentan abrirse camino en el espacio alrededor del metal. Una consecuencia interesante de este hecho es que muchos complejos que contienen fosfina no poseen 18 electrones de valencia. Los ejemplos incluyen Pt (PCy 3) 2, Pd [P (t-Bu) 3] 2 y [Rh (PPh 3) 3] +. ¿Eso no te vuelve loco? También vuelve locos a los complejos, y la mayoría de estos compuestos coordinadamente insaturados son catalizadores maravillosos.
El puenteo por fosfinas es extremadamente raro, pero los ligandos que contienen múltiples donantes de fosfina que se unen de manera L n (n > 1) a un solo centro metálico están por todas partes. Estos ligandos se denominan quelantes o polidentados para indicar que se enganchan a centros metálicos a través de múltiples sitios de unión. Por razones entrópicas, los ligandos quelantes se unen a un solo centro metálico en múltiples puntos si es posible, en lugar de unirse a dos centros metálicos diferentes (el efecto quelato acertadamente llamado). Una característica importante de las fosfinas quelantes es el ángulo de mordida, definido como el ángulo P—M-P predominante en complejos conocidos del ligando. Más adelante entraremos en los interesantes efectos del ángulo de mordida, pero por ahora, podríamos imaginar lo “infeliz” que sería un ligando con un ángulo de mordida preferido de 120° en la geometría plana cuadrada. Preferiría mucho ser parte de un complejo bipiramidal trigonal, por ejemplo.
La interacción orbital predominante que contribuye a la unión de fosfina es la que esperamos, un par solitario sobre fósforo interactuando con un orbital d metálico vacío. La naturaleza electrónica de los grupos R influye en la capacidad donadora de electrones del átomo de fósforo. Por ejemplo, las alquilfosfinas, que poseen enlaces P—Csp 3, tienden a ser mejores donadores de electrones que las arilfosfinas, que poseen enlaces P—Csp 2. La justificación aquí es la mayor electronegatividad del híbrido orbital sp 2 versus el híbrido sp3, lo que hace que el átomo de fósforo se mantenga más firmemente a su par solitario cuando se une a un carbono sp 2. La misma idea se aplica cuando los grupos aceptores y donadores de electrones se incorporan a R: la densidad de electrones en P es baja cuando R contiene grupos aceptores de electrones y alta cuando R contiene grupos donadores de electrones. Los ligandos (y los metales asociados) en la primera clase se denominan pobres en electrones, mientras que los de la segunda clase son ricos en electrones.
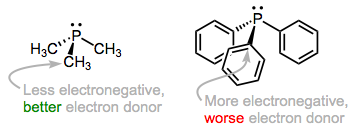
A medida que agregamos grupos R electronegativos, el átomo de fósforo (y el metal al que está unido) se vuelven más pobres en electrones.
Al igual que el CO, las fosfinasparticipan en el backbonding hasta cierto punto; sin embargo, el fenómeno aquí es de una naturaleza fundamentalmente diferente a la del backbonding de CO. Por un lado, las fosfinas carecen de un orbital π*. En los días de antaño, los químicos atribuyeron el backbonding en complejos de fosfina a una interacción entre un orbital d π metálico y un orbital 3d vacío sobre fósforo. Sin embargo, esta idea ha sido elegantemente probada como falso, y una explicación mucho más amigable con los organiceros ha tomado su lugar (¡no se requieren d orbitales en P!). En una serie de experimentos iluminantes, las longitudes de enlace M—P y P—R se midieron mediante cristalografía para varios pares redox de complejos. He elegido dos ejemplos ilustrativos, aunque la referencia enlazada está repleta de otros pares. La pregunta es: ¿cómo explicamos los cambios en la longitud del enlace tras la oxidación?
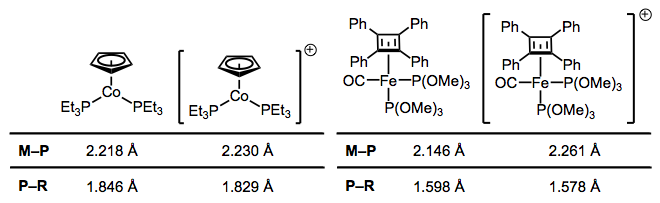
Tras la oxidación, las longitudes de los enlaces M—P aumentan y disminuyen las longitudes de los enlaces P—R. ¿Por qué?
La oxidación disminuye la capacidad del metal para unirse por retroceso, ya que elimina la densidad de electrones del metal. Esto explica los aumentos en la longitud del enlace M—P, solo imagínese una disminución del orden de bonos M—P debido a un peor backbonding. ¿Y la disminución de la longitud del enlace P—R? Es importante ver que invocar solo los orbitales 3d de fósforo no explicaría los cambios en las longitudes de los enlaces P-R, ya que los orbitales atómicos 3d están localizados definitivamente en el fósforo. En cambio, debemos invocar la participación de orbitales σ*P—R en el backbonding de fosfina para dar cuenta de las disminuciones de longitud P-R. Cuando todo está dicho y hecho, el LUMO de la fosfina libre tiene en su mayoría carácter antiadherentes P—R, con algunos 3d arrojados a la mezcla. La siguiente figura representa una de las interacciones involucradas en el backbonding M—P, una interacción d π → σ* (una interacción ortogonal d π → σ* también juega un papel). Al igual que con el CO, una estructura de resonancia que representa un doble enlace M=P es una heurística útil! Naturalmente, los grupos R que son más capaces de estabilizar la carga negativa, es decir, los grupos aceptores de electrones, facilitan el backbonding en las fosfino. Los metales ricos en electrones también ayudan.
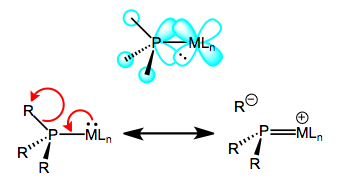
Backbonding en fosfinas, un asunto de ruptura de enlaces sigma.
Las propiedades estéricas y electrónicas de las fosfinasvarían enormemente. Tolman ideó algunos parámetros intrigantes que caracterizan las propiedades estéricas y electrónicas de esta clase de ligandos. Para abordar los esteros, desarrolló la idea del ángulo de cono, el ángulo del vértice de un cono formado por un punto 2.28 Å del átomo de fósforo (una longitud idealizada de enlace M—P), y los bordes más externos de los átomos en los grupos R, cuando los grupos R se doblan lo más posible hacia atrás. Los ángulos de cono más amplios, razonó Tolman, indican una mayor congestión estérica alrededor del átomo de fósforo. Para abordar la electrónica, Tolman utilizó a un amigo no tan antiguo: la frecuencia de estiramiento de CO (NCO) de los complejos mixtos de fosfina-carbonilo. Específicamente, utilizó complejos de Ni (CO) 3L, donde L es una fosfina terciaria, como su estándar. ¿Puedes anticipar la lógica de Tolman? ¿Cómo debería cambiar el NCO a medida que aumenta la capacidad de donación de electrones de los ligandos de fosfina?
La lógica de Tolman fue la siguiente: las fosfinas donadoras de electrones más fuertemente se asocian con más metales ricos en electrones, que son mejores en la unión posterior de CO (debido fundamentalmente a energías orbitales más altas). Un mejor backbonding de CO corresponde a un menor NCO debido a la disminución del orden de enlace C—O. Por lo tanto, los mejores ligandos donadores deberían estar asociados con valores menores de NCO (y viceversa para los ligandos aceptores de electrones). ¿Estaba en lo cierto? Exposicion A...
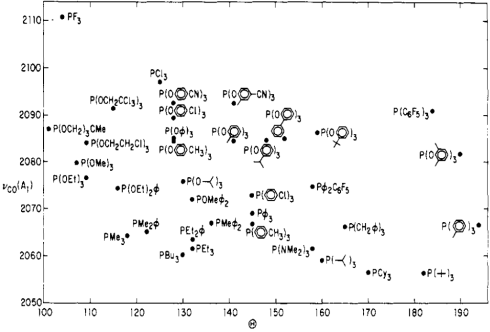
Mapa de Tolman de las propiedades estéricas y electrónicas de los ligandos de fosfina.
Observe el pobre ligando trifluorofosfina pegado en la esquina “muy pequeña, muy retiradora”, y su opuesto, la gigantesca tri (terc-butil) fosfina en la esquina “extremadamente voluminosa, muy donadora”. ¡Intrigante! Uno puede aprender mucho con solo estudiar esta tabla.
Dr. Michael Evans (Georgia Tech)
Adhesión en Complejos de Fosfina
Echemos un vistazo al diagrama MO de la\(\ce{PH3}\) molécula y comparémoslo con la\(\ce{NH3}\) molécula. Como cabría esperar, los MO son en general similares, pero hay una diferencia importante. Mientras que el LUMO in\(\ce{NH3}\) es el orbital anti-unión 3a 1, los LUMO en la\(\ce{PH3}\) molécula son los orbitales 2e antienlace. Las energías relativas de los orbitales 3a 1 y 2e en las\(\ce{NH3}\) moléculas\(\ce{PH3}\) y se intercambian hacia arriba. Esto puede atribuirse al hecho de que el átomo P utiliza los orbitales 3s y 3p como orbitales de valencia, mientras que N usa los orbitales 2s y 2p. Los orbitales 3s y 3p son más grandes y se superponen menos eficazmente con los orbitales pequeños 1s del hidrógeno. También tienen una mayor energía haciendo que los enlaces P-H sean menos polares que los enlaces N-H. La energía del\(\ce{PH3}\) HOMO es mayor que la del\(\ce{NH3}\) HOMO. Tanto el HOMO como el LUMO de\(\ce{PH3}\) son más difusos y polarizables que los orbitales respectivos en\(\ce{NH3}\).
La mayor energía del HOMO en lo\(\ce{PH3}\) convierte en un mejor donante que\(\ce{NH3}\). Además, el\(\ce{PH3}\) tiene propiedades\(\pi\) aceptoras debido a que los 2e LUMO son orbitales 2e antiadherentes relativamente bajos y tienen forma adecuada para\(\pi\) -solapamiento con orbitales d metálicos. \(\ce{NH3}\)no tiene estas propiedades\(\pi\) aceptoras porque su LUMO es el orbital 3a 1, y no los orbitales 2e. Los orbitales 2e\(\ce{NH3}\) son energéticamente demasiado altos para permitir interacciones\(\pi\) significativas-aceptor.
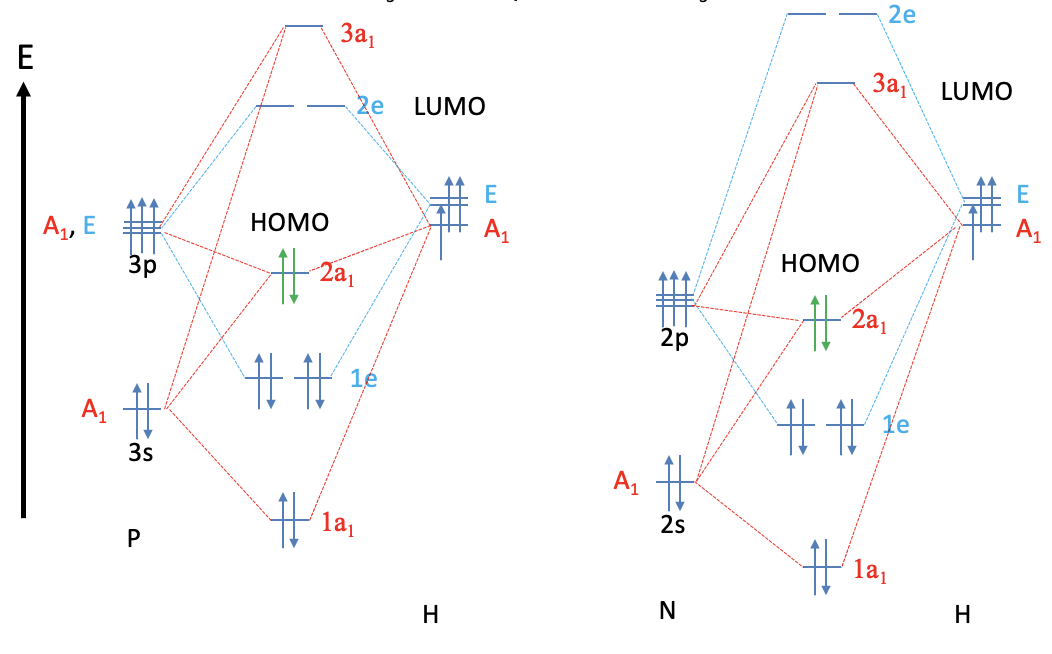
Las propiedades\(\sigma\)\(\pi\) donadoras y aceptoras del ligando de fosfina pueden modificarse sustituyendo H por otros grupos. Generalmente, más grupos donantes de electrones incrementan la energía del HOMO y del LUMO. Esto fortalece al\(\sigma\) donante y reduce las propiedades\(\pi\) -aceptoras. Viceversa, más grupos retiradores de electrones disminuyen la energía del HOMO y del LUMO. Como consecuencia, el ligando se convierte en un\(\sigma\) donador más débil y un\(\pi\) aceptor más fuerte (Fig. \(\PageIndex{4}\)).
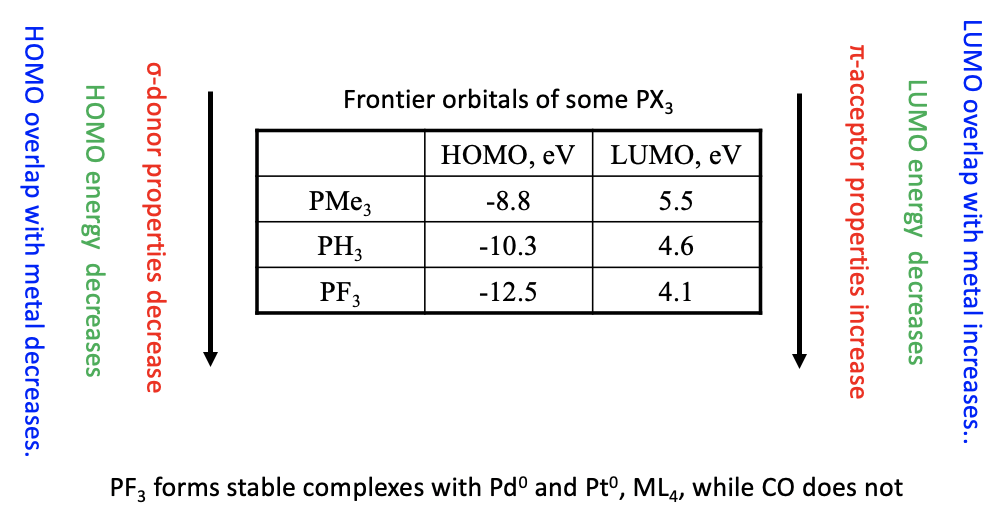
En la tabla anterior se muestran las energías HOMO y LUMO de tres fosfinas. Como era de esperar, las energías HOMO y LUMO disminuyen de\(\ce{PMe3}\)\(\ce{PH3}\) a\(\ce{PF3}\) debido a la naturaleza cada vez más atrayente de electrones del sustituyente. Como consecuencia, las propiedades\(\sigma\) -donadoras se debilitan y las propiedades\(\pi\) -aceptoras se fortalecen de a\(\ce{PMe3}\) a\(\ce{PH3}\) a\(\ce{PF3}\).
No solo las energías, sino también el solapamiento orbital es importante para la fuerza de las propiedades\(\pi\) aceptoras de los ligandos de fosfina. Cuanto más fuertemente atrayendo electrones es el grupo, más se localizan los orbitales LUMO de tipo e anti-enlace en el átomo P. Cuanto más se localicen estos orbitales en P, mejor se pueden solapar con el ligando. Viceversa la localización del HOMO se desplaza hacia el grupo a medida que el grupo se vuelve más atrayente de electrones, debilitando así las propiedades del\(\sigma\) donante.
Los ligandos de fosfina con grupos fuertemente aceptores de electrones tales como\(\ce{PF3}\) tienen propiedades\(\pi\) aceptoras lo suficientemente fuertes como para estabilizar los metales en bajos números de oxidación, similar a\(\ce{CO}\). Por ejemplo, el\(\ce{PF3}\) ligando forma complejos estables con átomos de Pd y Pt en el número de oxidación 0, mientras que no se conocen los respectivos carbonilos de Pd y Pt.