
15.6: Reacciones que involucran el enlace C-O de alcoholes
- Page ID
- 73012

Propiedades electrofílicas de los alcoholes
La formación de haluro de alquilo a partir de un alcohol y un haluro de hidrógeno proporciona un ejemplo importante de una reacción en la que se rompe el\(\ce{C-O}\) enlace del alcohol:
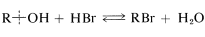
La reacción es reversible y la dirección favorecida depende de la concentración de agua. Los bromidos primarios a menudo se preparan mejor pasando bromuro de hidrógeno seco en el alcohol calentado a un poco por debajo de su punto de ebullición.
La formación de haluros se produce a una velocidad útil solo en presencia de ácido fuerte, que puede ser proporcionado por exceso de bromuro de hidrógeno o, generalmente y de manera más económica, por ácido sulfúrico. El alcohol acepta un protón del ácido para dar un ion alquiloxonio, el cual es más reactivo en el desplazamiento posterior con el ion bromuro que el alcohol (por cualquiera\(S_\text{N}2\) o\(S_\text{N}1\) mecanismos) debido a que\(\ce{H_2O}\) es un mejor grupo lábil que\(\ce{OH}^\ominus\):
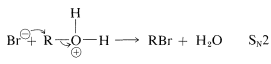
o
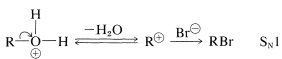
Si la reacción de desplazamiento es un\(S_\text{N}2\) proceso\(S_\text{N}1\) o depende de la estructura del alcohol. En general, se considera que los alcoholes primarios reaccionan por\(S_\text{N}2\) y los alcoholes secundarios y terciarios por\(S_\text{N}1\) mecanismos.
El cloruro de hidrógeno es menos reactivo que el bromuro de hidrógeno hacia los alcoholes primarios, y es posible que se requiera un catalizador (cloruro de zinc). Una solución de cloruro de zinc en ácido clorhídrico concentrado (reactivo Lucas) es un reactivo conveniente para diferenciar entre alcoholes primarios, secundarios y terciarios con menos de ocho carbonos aproximadamente. Los alcoholes terciarios reaccionan muy rápidamente para dar una capa insoluble de cloruro de alquilo a temperatura ambiente. Los alcoholes secundarios reaccionan en varios minutos, mientras que los alcoholes primarios forman cloruros solo al calentarse. El orden de reactividad es típico de\(S_\text{N}1\) las reacciones. El cloruro de zinc probablemente ayuda a romper el\(\ce{C-O}\) enlace del alcohol tanto como el ion plata ayuda a la ionización de\(\ce{RCl}\) (Sección 8-7D):
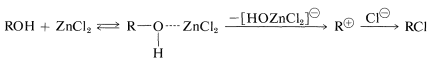
El cloruro de tionilo\(\ce{O=SCl_2}\),, es útil para la preparación de cloruros de alquilo, especialmente cuando no es deseable el uso de reactivos fuertemente ácidos, tales como cloruro de zinc y ácido clorhídrico. El cloruro de tionilo puede considerarse como el cloruro de ácido del ácido sulfuroso\(\ce{O=S(OH)_2}\), y como la mayoría de los cloruros de ácido, el halógeno es desplazado fácilmente por los alcoholes. La adición de 1 mol de un alcohol a 1 mol de cloruro de tionilo da un clorosfito de alquilo inestable, que generalmente se descompone con calentamiento suave para producir el cloruro de alquilo y el dióxido de azufre:
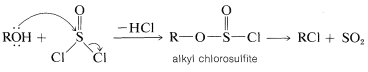
Los cloruros se pueden preparar de esta manera a partir de alcoholes primarios y secundarios, pero no terciarios. En la práctica, se agrega un equivalente de una base débil, como piridina (azabenzeno), para neutralizar el cloruro de hidrógeno que se forma. Si no se elimina el ácido, pueden ocurrir reacciones indeseables de degradación, eliminación y reordenamiento.
La reacción del cloruro de tionilo aparentemente puede proceder de la etapa de clorosulfito de alquilo por más de un mecanismo: una reacción en\(S_\text{N}2\) cadena iónica con ion cloruro,
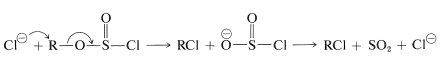
o una ionización\(S_\text{N}1\) similar y colapso del par\(\ce{R}^\oplus \ce{Cl}^\ominus\) iónico resultante para dar\(\ce{RCl}\):
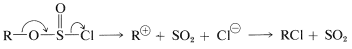
o
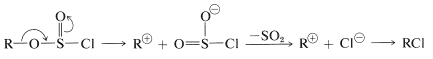
Obviamente, cuanto mayor sea la\(S_\text{N}2\) reactividad asociada con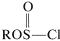 mejor irá la\(S_\text{N}2\) reacción y, a la inversa, si\(\ce{R}^\oplus\) se forma fácilmente a partir de la\(S_\text{N}1\) reacción es probable que se vea favorecida.
mejor irá la\(S_\text{N}2\) reacción y, a la inversa, si\(\ce{R}^\oplus\) se forma fácilmente a partir de la\(S_\text{N}1\) reacción es probable que se vea favorecida.
Otros haluros que son útiles en la conversión de alcoholes en haluros de alquilo son\(\ce{PCl_5}\)\(\ce{PCl_3}\),\(\ce{PBr_3}\), y\(\ce{PI_3}\), que son haluros de ácido de oxiácidos de fósforo. Al igual que con el cloruro de tionilo, a menudo se usa una base débil para facilitar la reacción. La base actúa para neutralizar el ácido formado, y también para generar iones bromuro para\(S_\text{N}\) las reacciones:
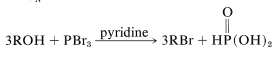
Ésteres de sulfato y sulfonato
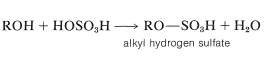
Es posible preferir ésteres de ácido sulfúrico por la reacción de un alcohol con el ácido:
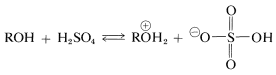
La reacción está estrechamente relacionada con la formación de haluros de alquilo en condiciones fuertemente ácidas, por lo que la conversión del alcohol en una sal de oxonio es una primera etapa:
La conversión del hidrogenosulfato de oxonio en el éster probablemente procede por un\(S_\text{N}2\) mecanismo con alcoholes primarios y un\(S_\text{N}1\) mecanismo con alcoholes terciarios:
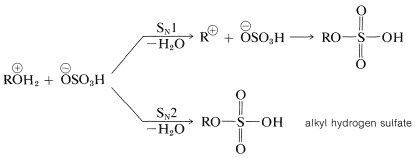
Un mecanismo alternativo, que opera en\(100\%\), o en ácido sulfúrico fuminante (que contiene disuelto\(\ce{SO_3}\)), es la adición de trióxido de azufre al\(\ce{OH}\) grupo:
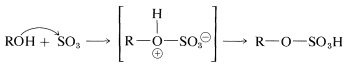
Las sales de sodio de los ésteres de alquil hidrogenosulfato tienen propiedades útiles como detergentes si el grupo alquilo es grande,\(\ce{C_{12}}\) o así:

El mecanismo de acción detergente será considerado con más detalle en el Capítulo 18.
En principio, los sulfatos de dialquilo podrían formarse por una\(S_\text{N}2\) reacción entre una sal de alquiloxonio y un ion alquilsulfato:
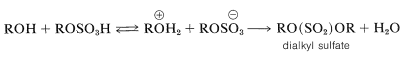
De hecho, si el metanol se calienta con ácido sulfúrico fumiante\(\ce{CH_3O(SO_2)OCH_3}\), se obtiene sulfato de dimetilo,, pero otros alcoholes se convierten mejor en sulfatos de dialquilo por oxidación de los sulfitos de dialquilo correspondientes formados por la reacción de 1 mol de cloruro de tionilo\(\left( \ce{SOCl_2} \right)\) con 2 moles del alcohol:
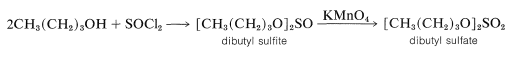
La razón por la que los sulfatos de dialquilo rara vez se preparan por reacción directa del alcohol con\(\ce{H_2SO_4}\) es que los monoésteres reaccionan rápidamente al calentarse para eliminar el ácido sulfúrico y formar alquenos, como se explica en la Sección 15-5C.
Los ácidos sulfónicos,\(\ce{R-SO_2-OH}\) o\(\ce{Ar-SO_2-OH}\), son oxiácidos de azufre que se asemejan al ácido sulfúrico\(\ce{HO-SO_2-OH}\), pero en los que el azufre se encuentra en un estado de oxidación inferior.
Los ésteres de sulfonato son intermedios útiles en reacciones de desplazamiento (Sección 8-7C) y proporcionan una ruta para la conversión de un alcohol\(\ce{ROH}\), a,\(\ce{RX}\) por la secuencia:
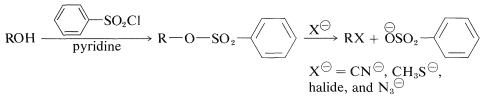
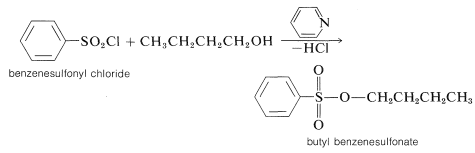
Los ésteres de sulfonato generalmente se preparan a través del tratamiento del alcohol con el cloruro de ácido (cloruro de sulfonilo) en presencia de piridina (azabenceno):
Deshidratación de Alcoholes con Ácidos Fuertes
En la reacción de un alcohol con ácido sulfúrico concentrado caliente, el alcohol se deshidrata a un alqueno:
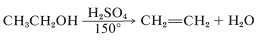
Esto es lo contrario de la hidratación catalizada por ácido de los alquenos discutida anteriormente (Sección 10-3E) y va a completarse si se deja que el alqueno destile de la mezcla de reacción a medida que se forma. Un mecanismo de deshidratación implica la transferencia de protones del ácido sulfúrico al alcohol, seguido de una\(E2\) reacción del ion sulfato de hidrógeno o agua con la sal oxonio del alcohol:
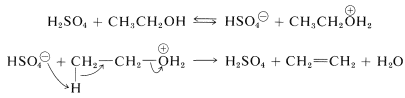
Alternativamente, el hidrogenosulfato de alquilo podría formarse y eliminar el ácido sulfúrico mediante una\(E2\) reacción:
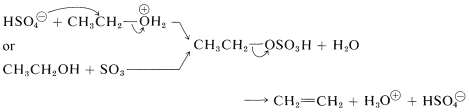
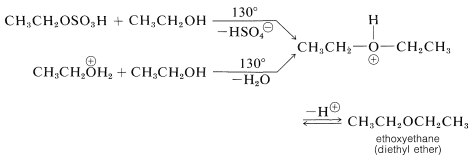
A temperaturas más bajas la sal de oxonio o el hidrogenosulfato de alquilo pueden reaccionar por un mecanismo de\(S_\text{N}\) desplazamiento con exceso de alcohol en la mezcla de reacción, formando así un éter dialquílico. Aunque cada etapa en la reacción es reversible, la formación de éter se puede mejorar destilando el éter tan rápido como se forma. El éter dietílico se fabrica comercialmente mediante este procedimiento:
La mayoría de los alcoholes también se deshidratarán a temperaturas bastante altas en presencia de catalizadores sólidos como gel de sílice u óxido de aluminio para dar alquenos o éteres. El comportamiento del etanol es razonablemente típico de los alcoholes primarios y se resume en las siguientes ecuaciones:
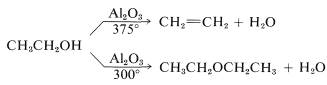
\(\ce{C-O}\)Escisión de enlaces de alcoholes terciarios
Los alcoholes terciarios reaccionan con el ácido sulfúrico a temperaturas mucho más bajas que la mayoría de los alcoholes primarios o secundarios. Las reacciones son típicamente\(S_\text{N}1\) y\(E1\) por medio de un carbocatión terciario, como se muestra aquí para el alcohol terc - butílico y el ácido sulfúrico:
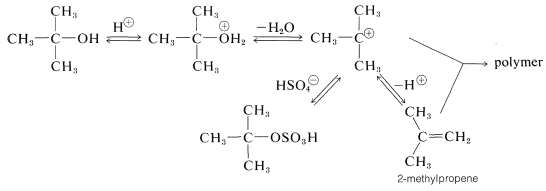
El 2-metilpropeno se puede eliminar de la mezcla de reacción por destilación y fácilmente se convierte en el producto principal mediante el ajuste apropiado de las condiciones de reacción. Si el 2-metilpropeno no se elimina a medida que se forma, el polímero y los productos de oxidación adquieren importancia. El ácido sulfúrico a menudo es un reactivo indebidamente extenuante para la deshidratación de alcoholes terciarios. El hidrogenosulfato de potasio, el sulfato de cobre, el yodo, el ácido fosfórico o el pentóxido de fósforo pueden dar mejores resultados al causar menos polimerización y menor degradación oxidativa que, con ácido sulfúrico, resulta en la formación de dióxido de azufre.
El\(E1\) comportamiento\(S_\text{N}1\) - de los alcoholes terciarios en ácidos fuertes se puede utilizar de manera beneficiosa en la preparación de éteres terc - butílicos. Si, por ejemplo, una mezcla de alcohol terc - butílico y metanol se calienta en presencia de ácido sulfúrico, el alcohol terciario reacciona rápida pero reversiblemente para producir 2-metilpropeno por medio del catión terc- butilo. Este catión puede ser atrapado por el metanol para formar terc - butil metil éter. Se pueden obtener altos rendimientos de éteres de esta manera:
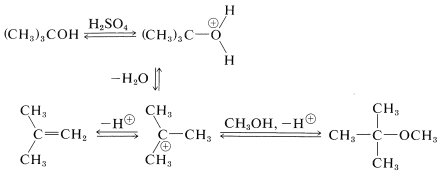
Reordenamientos de carbocationes
El reordenamiento de los grupos alquilo de los alcoholes es muy común en la deshidratación, particularmente en presencia de ácidos fuertes, que conducen a la formación de carbocationes. Ejemplos típicos que muestran tanto la migración de metilo como de hidrógeno siguen:
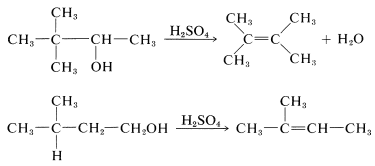
El paso clave en cada reordenamiento es la isomerización de un carbocatión, como se discute en la Sección 8-9B. Bajo control cinético, los productos finales siempre corresponden al reordenamiento de un carbocatión menos estable a un carbocatión más estable. (El control termodinámico puede llevar a resultados muy diferentes, Sección 10-4A.)
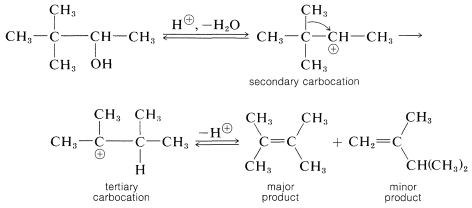
En la deshidratación del 3,3-dimetil-2-butanol, inicialmente se forma un carbocatión secundario, el cual se reorganiza a un carbocatión terciario cuando un grupo metilo vecino con su par de electrones de enlace migra al carbono positivo. La carga se transfiere así al carbono terciario:
Ésteres de Fosfato
El ácido fosfórico\(\left( \ce{H_3PO_4} \right)\) a menudo se usa en lugar del ácido sulfúrico para deshidratar alcoholes. Esto se debe a que el ácido fosfórico es menos destructivo; es tanto un ácido más débil como un agente oxidante menos potente que el ácido sulfúrico. La deshidratación probablemente procede por mecanismos similares a los descritos para el ácido sulfúrico (Sección 15-5C) y muy probablemente a través de la formación intermedia de un éster fosfato:
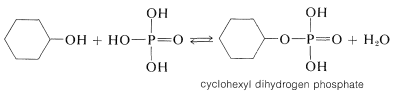
El éster puede eliminar\(\ce{H_3PO_4}\), como los ésteres de sulfato eliminan\(\ce{H_2SO_4}\), para dar alquenos:
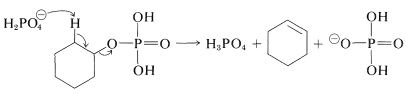
La química de los ésteres de fosfato es más complicada que la de los ésteres de sulfato porque es posible tener uno, dos o tres grupos alquilo sustituidos por los hidrógenos ácidos del ácido fosfórico:
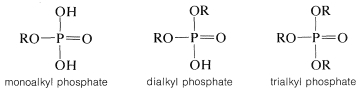
Además, el ácido fosfórico forma una extensa serie de anhidridos (con\(\ce{P-O-P}\) enlaces), que diversifican aún más el número y tipo de ésteres de fosfato. Los ésteres fosfatos más importantes son los derivados del ácido mono-, di- y trifosfórico (a veces clasificados como ácidos orto-, piro- y meta-fosfórico, respectivamente):
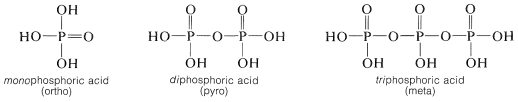
El equilibrio entre los ésteres de cualquiera de estos ácidos fosfóricos y el agua favorece la hidrólisis:
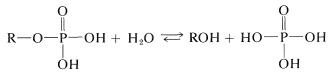
Sin embargo, los ésteres de fosfato son lentos para hidrolizarse en agua (a menos que esté presente un catalizador). La diferencia en la estabilidad cinética y termodinámica de los ésteres de fosfato hacia la hidrólisis se utiliza con gran efecto en sistemas biológicos.
De particular importancia es la conversión de gran parte de la energía que resulta de la fotosíntesis, o de la oxidación de grasas, carbohidratos y proteínas en las células en la formación de enlaces éster fosfato\(\left( \ce{C-O-P} \right)\) o enlaces anhídrido fosfato\(\left( \ce{P-O-P} \right)\). La energía así almacenada se utiliza en otras reacciones, cuyo resultado neto es la hidrólisis:
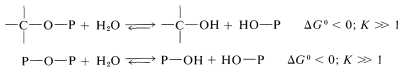
La sustancia que es la fuente inmediata de energía para muchas reacciones biológicas es el trifosfato de adenosina (ATP). Si bien se trata de una molécula bastante grande y compleja, el fin de negocio para el propósito de esta discusión es el grupo trifosfato. La hidrólisis de este grupo puede ocurrir para dar adenosina difosfato (ADP), adenosina monofosfato (AMP) o adenosina misma:
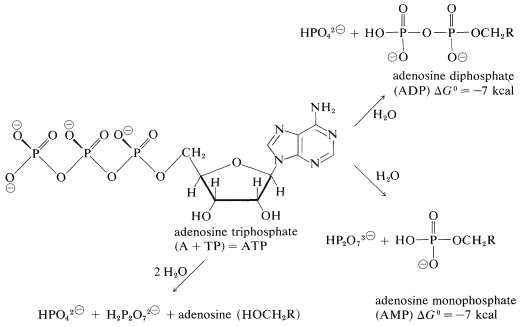
(Los grupos fosfato se representan aquí como la forma ionizada principal presente a pH\( \cong 7\) en soluciones de ATP).
Todas estas reacciones de hidrólisis son energéticamente favorables\(\left( \Delta G^0 < 0 \right)\), pero no ocurren directamente porque el ATP reacciona lentamente con el agua. Sin embargo, la hidrólisis del ATP es el resultado indirecto de otras reacciones en las que participa. Por ejemplo, como mostramos en la Sección 15-4D, el equilibrio para la formación directa de un éster a partir de un ácido carboxílico y un alcohol en fase líquida no es muy favorable (Ecuación 15-2). Sin embargo, si la esterificación puede acoplarse con la hidrólisis de ATP (Ecuación 15-3), la reacción global (Ecuación 15-4) se vuelve mucho más favorable termodinámicamente que la esterificación directa.
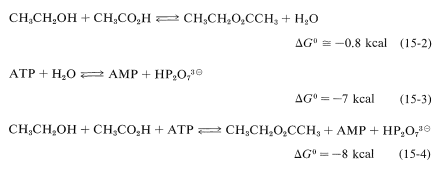
La hidrólisis de ATP podría acoplarse a la esterificación (u otras reacciones) de varias maneras. Lo más sencillo sería que la ATP convirtiera a uno de los participantes a un intermedio más reactivo. Para la esterificación, el intermedio reactivo es un derivado acilo de AMP formado por el desplazamiento de difosfato de ATP:
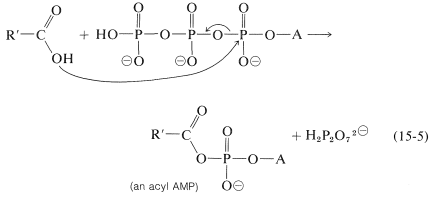
El acil AMP es como un cloruro de acilo,\(\ce{RCOCl}\), al tener un grupo lábil (AMP) que puede ser desplazado con un alcohol:
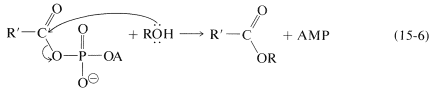
El resultado neto de la secuencia en las Ecuaciones 15-5 y 15-6 es la esterificación de acuerdo con la Ecuación 15-4. No es una esterificación catalizada porque en el proceso una molécula de ATP se convierte en AMP y difosfato por cada molécula de éster formada. El AMP tiene que ser reconvertido a ATP para volver a participar. Estas reacciones son llevadas a cabo por las células bajo la influencia catalítica de las enzimas. La parte adenosina de la molécula es crítica para la especificidad de acción de estas enzimas. Evidentemente, el funcionamiento de estas enzimas es de gran interés e importancia.
Si no te queda claro el papel de los ésteres de fosfato, como el ATP, en la realización de reacciones como la esterificación en medios acuosos bajo la influencia de enzimas en las células, piensa en cómo tratarías de llevar a cabo una esterificación de etanol en solución acuosa diluida. Recuerde que, con el agua en gran exceso, el equilibrio será bastante desfavorable para la reacción de esterificación de la Ecuación 15-2. Podría considerar agregar\(\ce{CH_3COCl}\), para lo cual el equilibrio para la formación de ésteres es mucho más favorable (Sección 15-4D). Sin embargo,\(\ce{CH_3COCl}\) reacciona violentamente con el agua para formar\(\ce{CH_3CO_2H}\), y esta reacción destruye el\(\ce{CH_3COCl}\) antes tiene muchas posibilidades de reaccionar con etanol para dar el éster. Claramente, lo que necesitarías es un reactivo que se\(\ce{CH_3CO_2H}\) convierta en algo que reaccione con etanol en agua para darle al éster una constante de equilibrio favorable y sin embargo no reaccionará muy rápido con el agua. Los ésteres de fosfato proporcionan esta función en sistemas bioquímicos al ser bastante poco reactivos al agua pero capaces de reaccionar con ácidos carboxílicos bajo la influencia de enzimas para dar acil fosfatos. Estos acil fosfatos pueden reaccionar entonces con alcoholes bajo la influencia de otras enzimas para formar ésteres en presencia de agua.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."