
21.7: ¿Cuál es mejor- MO o VB?
- Page ID
- 72822

La energía calculada del enlace electrón-par de la molécula de hidrógeno en función de la distancia\(\ce{H-H}\) intermolecular\(r\) por los procedimientos ab initio (exacto), MO y VB se muestra en la Figura 21-11. Los resultados muestran que ni los cálculos MO ni VB se acercan al cálculo ab initio en la reproducción de la energía de disociación experimental\(D_e\), o la variación de la energía con la distancia intermolecular. El método VB da un valor energético un poco mejor al mínimo y el método MO da malos resultados a valores mayores de\(r\). Podemos decir que, según se calcula por el método MO, la molécula no se “disocia adecuadamente”.
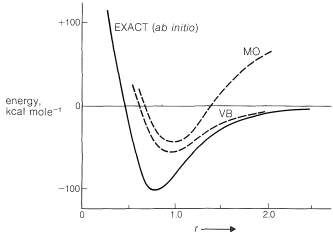
Dentro de los cálculos del método MO, la molécula no se “disocia adecuadamente”.
¿Por qué estos cálculos arrojan resultados tan alejados de la curva ab initio? Hay dos razones. En primer lugar, se utilizan orbitales atómicos que son apropiados para átomos aislados, pero difícilmente se espera que sean los mejores orbitales para los electrones cuando dos o más átomos están en estrecha proximidad. Es conveniente utilizar orbitales atómicos en cálculos simples porque son matemáticamente simples, pero se sabe que orbitales más complicados dan mejores resultados. Segundo, ninguno de los tratamientos toma debidamente en cuenta las repulsiones electrón-electrón. Para dos electrones, se requiere un término de la forma\(\frac{e^2}{r^2_{12}}\) (en el que\(e\)\(r_{12}\) está la carga electrónica y es la distancia entre los electrones) para describir la repulsión entre electrones. Los cálculos exactos evitan ambas dificultades pero son matemáticamente tan complejos que carecen de cualquier capacidad para proporcionar comprensión cualitativa.
El método VB da una energía ligeramente menor que el método MO como mínimo, ya que en el método MO simple, cuando calculamos la energía resultante de dos electrones que entran en el orbital molecular más bajo, no ponemos restricciones en que estén cerca. Como resultado, existe la\(50\%\) probabilidad de que ambos electrones estén simultáneamente en cualquiera de las dos mitades del orbital molecular. En contraste, el método simple VB combina configuraciones\(1\) y\(2\), cada una teniendo solo un electrón por orbital atómico, y no se tiene en cuenta la posibilidad de que ninguno de los orbitales atómicos contenga más de un electrón. Esto equivale a descuidar los esquemas de emparejamiento\(\ce{H}^\ominus \ce{H}^\oplus \leftrightarrow \ce{H}^\oplus \ce{H}^\ominus\). Ni la aproximación VB ni la MO son las mejores posibles; el método MO simple tiende a tener muy poco en cuenta la repulsión interelectrónica, mientras que el método VB tiende a tomar demasiado en cuenta la misma. Sin embargo, como puede verse en la Figura 21-11, tomar demasiado en cuenta la repulsión de electrones es la mejor aproximación.
¿Por qué un enlace de par de electrones calculado por el método MO no se disocia adecuadamente? Hemos visto que la mitad de las veces ambos electrones en el orbital molecular de baja energía están en las proximidades de solo uno de los núcleos. Pero a medida que los núcleos se alejan, esto corresponde a una energía mucho mayor que tener un solo electrón en las proximidades de cada núcleo, como sugiere el método VB.
No hay una respuesta inequívoca a la pregunta de cuál es el mejor método. Es probable que los cálculos por el método VB sean más confiables que los del método MO, pero en la práctica son mucho más difíciles de llevar a cabo. Para muchas moléculas de electrones, el procedimiento MO es más sencillo de visualizar porque combinamos orbitales atómicos en orbitales moleculares y luego poblamos los orbitales de menor energía con electrones. En el método VB, los orbitales atómicos están ocupados, pero los electrones de diferentes átomos se emparejan para formar enlaces, un proceso que requiere una consideración explícita de las funciones de ondas de muchos electrones. Por decirlo de otra manera, es más fácil visualizar un sistema de orbitales moleculares que contienen\(N\) electrones que visualizar una función de onda híbrida de\(N\) electrones.
¿Cómo se pueden mejorar los métodos MO y VB? La respuesta depende de lo que uno quiera: cálculos más precisos o mejor comprensión cualitativa. Para mejorar los cálculos de VB necesitamos orbitales que permitan que los electrones se extiendan sobre más de un átomo. Los orbitales GVB discutidos en la Sección 6-6 se ajustan a este propósito y dan una curva de energía solo ligeramente por encima de la curva exacta de la Figura 21-11. En el tratamiento GVB los orbitales se deslocalizan menos a medida que\(r\) aumenta.
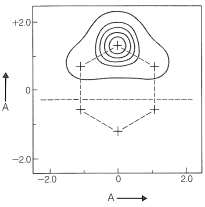
Cuando los orbitales atómicos se derivan por cada carbono del sistema\(\pi\) -electrónico del benceno por el método GVB, están algo más dispersos que\(p\) los orbitales simples de carbono (Figura 21-12). El uso de estos orbitales en los cálculos VB da excelentes resultados con solo los dos esquemas de emparejamiento de benceno,\(9\) y\(10\).
La mejora del método MO implica mejores orbitales, mejor explicación de la repulsión interelectrónica e introducción de mezcla de diferentes configuraciones de electrones en los orbitales moleculares (” interacción de configuración “). Los cálculos MO mejorados dan energías mucho más precisas al mínimo de una gráfica como la Figura 21-11, pero los enlaces aún no se disocian adecuadamente, por la misma razón que con el método MO simple.
No podemos decir que el método VB o MO sea más correcto; sólo que una aproximación puede ser más útil que la otra para intentar resolver un problema en particular. El hecho es que cuanto más se afina cada uno, más parecen fundirse en un procedimiento común; pero, desgraciadamente, en el proceso de refinamiento las matemáticas se vuelven tan complejas que la comprensión cualitativa de lo que se está haciendo tiende a desaparecer por completo.
No podemos decir que el método VB o MO sea más correcto; sólo que una aproximación puede ser más útil que la otra para intentar resolver un problema en particular.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."