
21.6: Aplicación de la Teoría MO a Otros Sistemas
- Page ID
- 72830

Moléculas polares
Muchas moléculas importantes tienen enlaces simples y dobles alternantes (están conjugadas), pero tienen átomos que son más (o menos) atrayentes de electrones que el carbono. Un ejemplo es propenal (acroleína),\(18\):
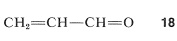
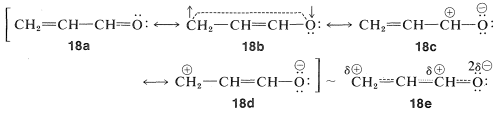
Con tales moléculas hay que tomar en cuenta el hecho de que los\(\pi\) electrones serán atraídos por el oxígeno del carbono, porque el oxígeno es más electronegativo que el carbono. Con el método VB podemos hacer esto considerando esquemas de emparejamiento de electrones iónicos,\(18c\) y\(18d\), junto con las estructuras diénicas,\(18a\) y\(18b\). El híbrido,\(18e\), se dibuja para reflejar las contribuciones relativas esperadas de las diversas formas,\(18a\) siendo de lo más importante.
Estructuras iónicas como\(19a\) y no\(19b\) necesitan ser consideradas para propenal porque el carbono es mucho menos atrayente de electrones que el oxígeno:
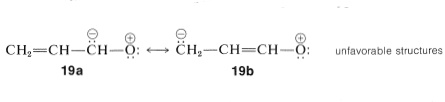
El análisis de la configuración electrónica resultante de los cálculos MO concuerda generalmente con el híbrido VB\(18e\).
El catión 2-propenilo (alilo)
Un tipo de carbocatión especialmente importante está representado por los esquemas de apareamiento de electrones de 2-propenilo\(21b\),\(21a\) y, que corresponden al híbrido\(21c\).
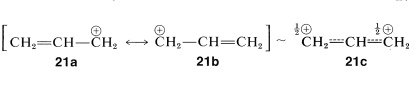
Debido a\(21b\) que\(21a\) y son equivalentes y no es posible otra estructura única de baja energía, se espera una energía de deslocalización considerable. La evidencia de esta energía de deslocalización\(21c\) está disponible a partir de la facilidad comparativa de reacciones que implican la formación de intermedios de carbocationes. Un ejemplo está en\(S_\text{N}1\) las ionizaciones de haluros de alquenil y alquilo. La ionización\(\ce{CH_2=CHCH_2Br} \rightarrow \ce{CH_2=CHCH_2^+} + \ce{Br^+}\) procede más fácilmente que\(\ce{CH_3CH_2CH_2Br} \rightarrow \ce{CH_3CH_2CH_2^+} + \ce{Br^+}\) (para lo cual no es posible la deslocalización de\(\pi\) electrones).
El tratamiento MO del catión 2-propenilo comienza con el modelo atómico-orbital\(22\):
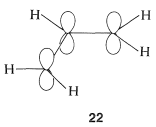
Cualquier\(\pi\) electrón será deslocalizado a través de los orbitales de\(22\), pero no es tan fácil estar seguro de que cuando dos electrones se colocan en el orbital molecular más bajo la distribución de electrones resultante será la misma que\(21c\) con la mitad de la carga positiva encendida\(\ce{C_1}\) y la mitad encendido\(\ce{C_3}\).
El cálculo completo da el resultado mostrado en la Figura 21-9. Aquí el orbital molecular de menor energía tiene una mayor proporción del\(p\) orbital de\(\ce{C_2}\) mezclado que los\(p\) orbitales de\(\ce{C_1}\) y\(\ce{C_3}\) - de hecho, la cantidad justa para tener\(\ce{C_2}\) neutro y\(\ce{C_1}\) y\(\ce{C_3}\) cada uno con\(\frac{1}{2}^\oplus\) cuando se llena este MO con dos electrones emparejados. La energía de deslocalización calculada para el catión es\(\left( 2 \alpha + 2.82 \beta \right) - \left( 2 \alpha + 2 \beta \right) = 0.82 \beta\) o aproximadamente\(16 \: \text{kcal}\) si\(\beta\) se toma para ser\(19 \: \text{kcal}\). Así, en todos los aspectos, los métodos simples VB y MO dan la misma representación del carbocatión 2-propenilo.
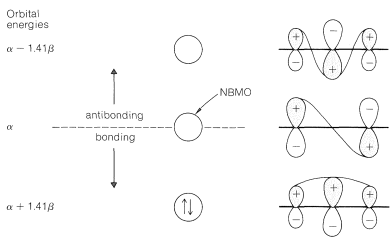
Notarás que el radical 2-propenilo y el carbanión 2-propenilo pueden formularse mediante el mismo conjunto de orbitales\(\pi\) moleculares (Figura 21-9) utilizados para el carbocatión poniendo uno o dos electrones en el MO no enlazado. Las energías de deslocalización calculadas para el radical y el anión son las mismas que para el catión. Así\(\left( 3 \alpha + 2.82 \beta \right) - \left( 3 \alpha + 2 \beta \right) = 0.82 \beta\) para el radical y\(\left( 4 \alpha + 2.82 \beta \right) - \left( 4 \alpha + 2 \beta \right) = 0.82 \beta\) para el anión.
Espectros Electrónicos por el Método MO
La sección 9-9B cubre explicaciones cualitativas de cómo se utiliza el método VB para dar cuenta de la radiación de menor energía (longitud de onda más larga) requerida para la excitación electrónica de los polienos conjugados en comparación con los polienos no conjugados. Así, el 1,3-butadieno tiene un\(\lambda_\text{max}\) para la luz ultravioleta at\(217 \: \text{nm}\), mientras que el 1,5-hexadieno tiene un\(\lambda_\text{max}\) at correspondiente\(185 \: \text{nm}\).
Ahora consideraremos cómo se puede utilizar el enfoque MO para comprender estas diferencias en la energía de excitación. Los niveles\(\pi\) de energía y las configuraciones electrónicas para 1,3-butadieno deslocalizado y localizado se muestran en la Figura 21-10 (ver también Sección 21-4). Debido a que los dobles enlaces están tan separados, el sistema\(\pi\) -electrón del 1,5-hexadieno por el enfoque MO simple es idéntico al del 1,3-butadieno localizado. El cambio de energía calculado para la\(\pi \rightarrow \pi^*\) transición de energía más baja es\(\left( \alpha - 0.62 \beta \right) - \left( \alpha + 0.6 \beta \right) = -1.24 \beta\) para 1,3-butadieno y\(\left( \alpha - \beta \right) - \left( \alpha + \beta \right) = -2 \beta\) para 1,5-hexadieno. En cada caso la energía del electrón en el\(\pi\) orbital ocupado más alto (el orbital HOMO) se resta de la energía que tendría un electrón en el orbital desocupado más bajo (el\(\pi^*\) orbital LUMO). Otras transiciones son posibles, como de un electrón desde el orbital de energía ocupado más bajo\(\alpha + 1.62 \beta\) hasta el orbital más alto desocupado de energía\(\alpha - 1.62 \beta\), pero éstas tendrían energías mucho mayores.
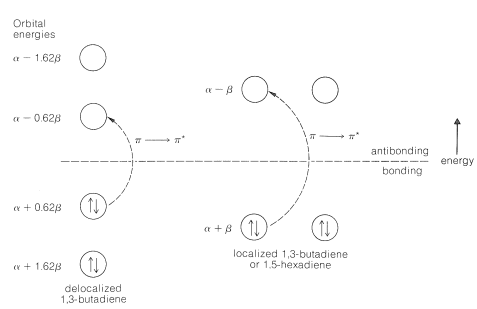
Cualitativamente, se predice que la energía de\(\pi \rightarrow \pi^*\) transición sea sustancialmente menor para 1,3-butadieno que para 1,5-hexadieno. Sin embargo, cualquier intento de correlación cuantitativa es sospechoso, ya que la\(\pi \rightarrow \pi^*\) transición energética más baja calculada para 1,3-butadieno es\(-1.24 \beta\) y, si\(\beta\) es\(19 \: \text{kcal}\) (ver Sección 21-3C),\(\lambda_\text{max}\) a partir de la Ecuación 9-2 debería ser\(1214 \: \text{nm}\) en lugar de la observada\(217 \: \text{nm}\).
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."