
22.3: Propiedades espectrales de Arenes
- Page ID
- 73594

Espectros Infrarrojos
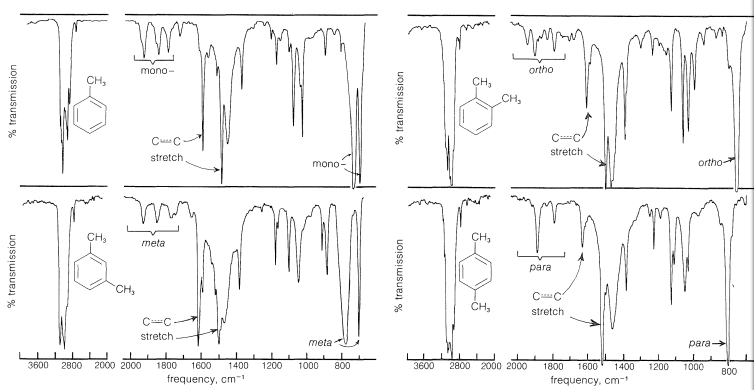
La presencia de un grupo fenilo en un compuesto se puede determinar con un grado de certeza razonable a partir de su espectro infrarrojo. Por ejemplo, en la Figura 22-1 vemos los espectros infrarrojos del metilbenceno, y del 1,2-, 1,3- y 1,4-dimetilbenceno. Que cada espectro es de un derivado de benceno es evidente a partir de ciertas características comunes. Las dos bandas cercanas\(1600 \: \text{cm}^{-1}\) y\(1500 \: \text{cm}^{-1}\), aunque de intensidad variable, se han correlacionado con las vibraciones de estiramiento de los enlaces carbono-carbono del anillo aromático; además, las bandas afiladas cercanas\(3030 \: \text{cm}^{-1}\) son características de\(\ce{C-H}\) los enlaces aromáticos. Otras bandas en los espectros, especialmente aquellas entre\(1650 \: \text{cm}^{-1}\) y\(2000 \: \text{cm}^{-1}\), entre\(1225 \: \text{cm}^{-1}\) y\(950 \: \text{cm}^{-1}\), y por debajo\(900 \: \text{cm}^{-1}\), se han correlacionado con el número y posición de los sustituyentes del anillo. Si bien no documentaremos todas estas diversas bandas en detalle, cada uno de los espectros de la Figura 22-1 está marcado para mostrar algunas de las correlaciones que se han realizado.
Espectros de absorción electrónica
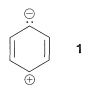
En comparación con los polienos conjugados de cadena lineal, los compuestos aromáticos tienen espectros de absorción relativamente complejos con varias bandas en la región ultravioleta. El benceno y los alquilbencenos muestran dos bandas en las que nos interesará primordialmente, una cerca\(200 \: \text{nm}\) y la otra cercana\(260 \: \text{nm}\). La\(\text{nm}\) banda\(200\) - es de intensidad bastante alta y corresponde a la excitación de un\(\pi\) electrón del sistema conjugado a un\(\pi^*\) orbital (es decir, una\(\pi \rightarrow \pi^8*\) transición). El estado excitado tiene contribuciones significativas de estructuras dipolares como\(1\):
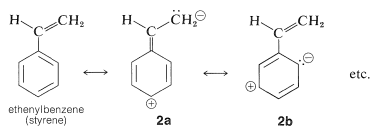
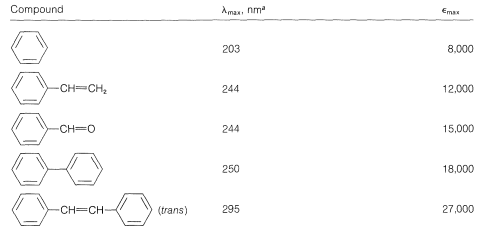
Esto es análogo a las bandas de absorción de dienos conjugados (Sección 9-9B) excepto que la longitud de onda de absorción de bencenos es más corta. De hecho, las\(200\) -\(\text{nm}\) absorciones de benceno y alquilbencenos están poco más allá del alcance de la mayoría de los espectrómetros de cuarzo comerciales. Sin embargo, estas absorciones (que decimos que surgen del cromóforo de benceno\(^2\)) se intensifican y se desplazan a longitudes de onda más largas cuando el sistema conjugado se extiende mediante la sustitución de los hidrógenos del anillo por grupos insaturados (por ejemplo\(\ce{-CH=CH_2}\),\(\ce{-C \equiv CH}\),\(\ce{-CH=O}\), y\(\ce{-C \equiv N}\); ver Cuadro 22-2). El sistema\(\pi\) de electrones deslocalizados del cromóforo absorbente ahora incluye los electrones del sustituyente insaturado así como los del anillo. En el caso específico del etenilbenceno el estado excitado es una estructura híbrida compuesta de\(2a\)\(2b\) y otras estructuras dipolares relacionadas:
Cuadro 22-2: Efecto de la conjugación sobre la absorción electrónica por el cromóforo de benceno
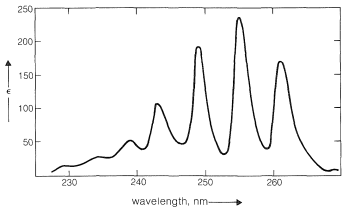
Se observan efectos similares para derivados de benceno en los que el sustituyente tiene pares de electrones no compartidos que pueden conjugarse con el anillo de benceno (por ejemplo,\(\ce{-NH_2}\),\(\ce{-OH}\),\(\ce{-Cl}\)). Un par de electrones no compartidos se deslocaliza hasta cierto punto para convertirse en parte del sistema de\(\pi\) electrones aromáticos tanto en el estado molido como en el estado excitado, pero lo que es más importante en el estado excitado. Esto se ilustra para la bencenamina (anilina) mediante las siguientes estructuras, que contribuyen a la estructura híbrida:
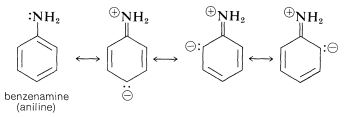
Los datos de la Tabla 22-3 muestran el efecto sobre el cromóforo de benceno de este tipo de sustituyente - el sustituyente a menudo se llama auxocromo. \(^2\)Este término significa que, aunque el sustituyente en sí no es responsable de la banda de absorción, desplaza la absorción del grupo cromóforo, en este caso el anillo de benceno, hacia longitudes de onda más largas. Los grupos auxocrómicos suelen aumentar la intensidad de la absorción también.
Cuadro 22-3: Efecto de los sustituyentes auxocrómicos sobre la absorción electrónica por el cromóforo de benceno

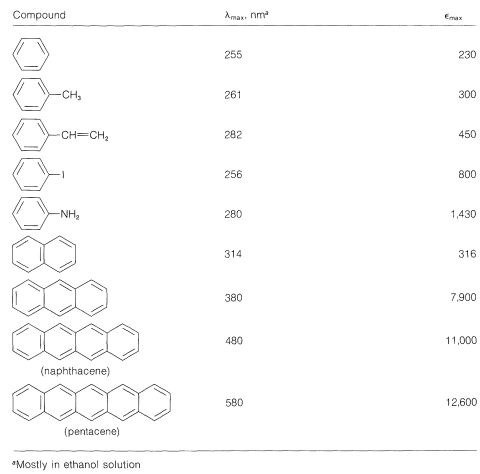
Cuadro 22-4: Efectos de la Estructura sobre la Absorción Electrónica Correspondiente a la Banda Benzenoide
La banda benzenoide corresponde a una\(\pi \rightarrow \pi^*\) transición de baja energía de las moléculas de benceno. La intensidad de absorción es débil porque el\(\pi^*\) estado involucrado tiene la misma simetría electrónica que el estado fundamental del benceno, y las transiciones entre estados simétricos suelen estar prohibidas. Las transiciones se observan en este caso solo porque las vibraciones del anillo hacen que se distorsione ligeramente en instantes dados. En el tratamiento de enlace de valencia este estado excitado del benceno es un estado antienlace de\(\pi\) los electrones.
Los espectros electrónicos de hidrocarburos aromáticos polinucleares como el naftaleno y el antraceno, en los que los anillos aromáticos se fusionan entre sí de manera lineal, se asemejan al espectro del benceno excepto que las bandas se desplazan a longitudes de onda más largas. De hecho, con los cuatro anillos linealmente conectados de naftaceno, la banda bencenoidea se desplaza lo suficientemente lejos a longitudes de onda más largas para estar en la región visible del espectro (ver Cuadro 22-4). En consecuencia, el naftaceno es amarillo. El siguiente miembro superior, el pentaceno, es azul.
Compuestos como fenantreno, criseno y pireno, en los que los anillos aromáticos se fusionan de manera angular, tienen espectros electrónicos complejos con considerable estructura fina. Los\(\lambda_\text{max}\) valores normalmente están a longitudes de onda más cortas que las de sus isómeros lineales.
Espectros de resonancia magnética nuclear
Los desplazamientos químicos de los protones de areno (\(6.5 \: \text{ppm}\)a\(8.0 \: \text{ppm}\)) son característicamente hacia campos magnéticos más bajos que los de los protones unidos a dobles enlaces ordinarios (\(4.6 \: \text{ppm}\)a\(6.9 \: \text{ppm}\)). La diferencia de aproximadamente\(2 \: \text{ppm}\) no se puede explicar fácilmente porque los hidrógenos en ambos tipos de sistemas están unidos al carbono a través\(sp^2\) de\(\sigma\) enlaces (Secciones 6-4C y 6-5A).
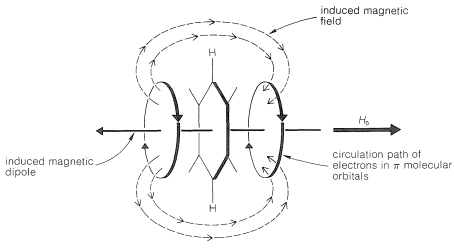
Al menos parte de la diferencia de desplazamiento químico entre protones de areno y protones alquenos es el resultado de la propiedad especial de\(\pi\) los electrones en sistemas aromáticos de circular libremente por encima y por debajo del plano de los núcleos de carbono, como se muestra en la Figura 22-4. Cuando una molécula como el benceno se somete a un campo magnético que tiene un componente perpendicular al plano del anillo, los electrones circulan alrededor del anillo de tal manera que producen un dipolo magnético local en la dirección opuesta al campo aplicado. Este efecto de blindaje diamagnético actúa para reducir el campo aplicado en el centro del anillo. Por lo tanto, si un protón pudiera ubicarse en el centro del anillo, el campo aplicado tendría que ser mayor de lo normal para contrarrestar el campo diamagnético local y llevar el protón a resonancia. Un protón fuera del anillo se ve afectado de manera opuesta (efecto deshielding paramagnético) porque, como puede verse en el diagrama, dichos protones se ubican en la trayectoria de retorno de las líneas de fuerza asociadas con el campo local y así se encuentran en un campo mayor que el que surge del externo imán solo. Cuando el plano de la molécula se orienta paralelo al campo, se corta la circulación diamagnética. Como resultado, a medida que las moléculas giran una y otra vez en el líquido, el componente de magnetización perpendicular al plano del anillo varía rápidamente. Sin embargo, los hidrógenos del anillo experimentan un efecto paramagnético neto sustancial. Por lo tanto, las posiciones de las líneas de resonancia se desplazan a campos magnéticos inferiores
Una fuerte evidencia en confirmación de la explicación anterior de los desplazamientos químicos de los hidrógenos aromáticos es proporcionada por un estudio del polieno conjugado cíclico [18] anuleno, que tiene hidrógenos tanto “dentro” como “fuera” del anillo:
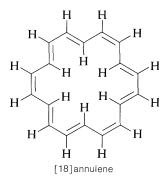
Los hidrógenos internos son fuertemente deshirados, llegando a\(1.9 \: \text{ppm}\) campo alto de tetrametilsilano, mientras que los hidrógenos externos son deshirados y vienen en\(8.8 \: \text{ppm}\) campo abajo de TMS. Como veremos, el efecto de la corriente del anillo es bastante general y constituye una prueba ampliamente utilizada para el carácter aromático en sistemas de anillos de polieno conjugado.
En general, las divisiones espín-espín observadas entre los cinco protones de un grupo fenilo pueden ser extremadamente complejas. Un ejemplo lo proporciona el nitrobenceno (Figura 22-5), que tiene diferentes desplazamientos químicos para sus hidrógenos orto, meta y para y seis constantes de interacción espín-espín diferentes:\(J_{23}\),,,\(J_{24}\),\(J_{25}\),\(J_{26}\),,\(J_{34}\),\(J_{35}\), (los subíndices corresponden a los números de posición del protones):
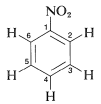
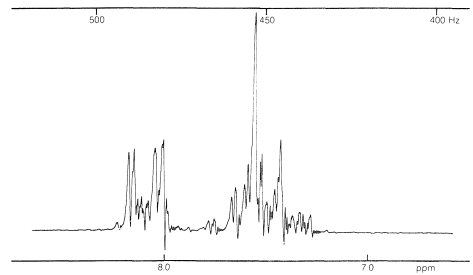
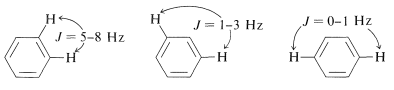
Tal espectro es demasiado complejo para ser analizado por cualquier procedimiento sencillo. Sin embargo, la resonancia magnética nuclear puede ser útil para asignar estructuras a derivados aromáticos, particularmente en conjunción con intensidades de línea integradas y valores aproximados de las constantes de acoplamiento entre los hidrógenos del anillo, como se muestra a continuación:
\(^2\)Un cromóforo es una agrupación de átomos en una molécula orgánica que da origen al color, o tiene el potencial de hacerlo cuando están presentes otros grupos llamados auxocromos (ver también la Sección 28-4).
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."