
22.4: Sustitución Aromática Electrofílica
- Page ID
- 73581

Alcance y Mecanismo
En esta sección nos interesarán principalmente las reacciones de los arenos que implican ataque a los átomos de carbono del anillo aromático. No profundizaremos ahora sobre las reacciones de los grupos sustituyentes alrededor del anillo.
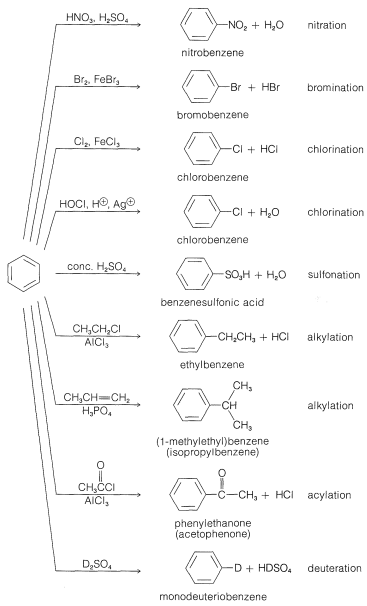
Los principales tipos de reacciones que involucran anillos aromáticos son la sustitución, adición y oxidación. De estos, el tipo más común es la sustitución electrofílica. En la Figura 22-7 se presenta un resumen de las reacciones de sustitución más importantes del benceno. Muchos de los reactivos utilizados para lograr estas sustituciones le resultarán familiares en relación con las reacciones de adición electrófila a los alquenos (por ejemplo\(\ce{Cl_2}\)\(\ce{Br_2}\),\(\ce{H_2SO_4}\),, y\(\ce{HOCl}\); Sección 10-3). La adición electrofílica a alquenos y la sustitución aromática electrófila son procesos polares, escalonados, y el paso clave para cada uno es el ataque de un electrófilo al carbono para formar un intermedio catiónico. Podemos representar este tipo de reacción mediante las siguientes ecuaciones generales, en las que el reactivo atacante se representa ya sea formalmente como un catión\(\ce{X}^\ominus\), o como una molécula neutra pero polarizada,\(\overset{\delta \oplus}{\ce{X}}\) —\(\overset{\delta \ominus}{\ce{Y}}\):
sustitución aromática electrofílica (primer paso)
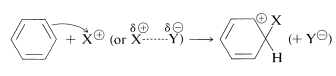
adición electrofílica a alquenos (primer paso)
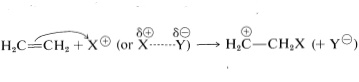
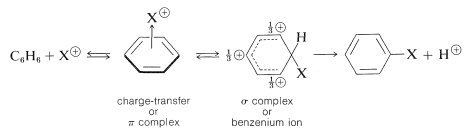
El intermedio mostrado para la sustitución aromática ya no tiene una estructura aromática; más bien, es un catión con cuatro\(\pi\) electrones deslocalizados sobre cinco núcleos de carbono, estando el sexto carbono saturado con enlaces\(sp^3\) híbridos. Puede formularse en términos de las siguientes estructuras contribuyentes, que se supone que contribuyen esencialmente por igual:
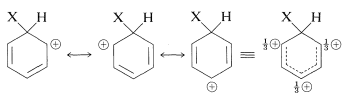
La importancia de escribir la estructura híbrida con las cargas parciales en estas tres posiciones se hará evidente más adelante. Este tipo de ion se conoce como un \(\sigma\)complejo o un ion benzenio.
El anillo aromático se regenera a partir de este intermedio catiónico por la pérdida de un protón del\(sp^3\) carbono hibridado. El par de electrones de este\(\ce{C-H}\) enlace se convierte entonces en parte del sistema\(\pi\) aromático-electrónico y se forma un producto de sustitución de benceno\(\ce{C_6H_5X}\),,.
sustitución aromática electrofílica (segunda etapa)
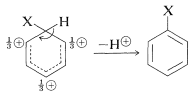
La ganancia en la estabilización asociada a la regeneración del anillo aromático es suficientemente ventajosa para que esto, en lugar de la combinación del catión con\(\ce{Y}^\ominus\), normalmente sea el curso de reacción favorecido. Aquí radica la diferencia entre la sustitución aromática y la adición de alquenos. En el caso de los alquenos generalmente no hay energía de resonancia sustancial que se pueda obtener por la pérdida de un protón del intermedio, que tiende por tanto a reaccionar por combinación con un reactivo nucleofílico.
adición electrófila a alquenos (segunda etapa)
\[\overset{\oplus}{\ce{C}} \ce{H_2-CH_2X} + \ce{Y}^\ominus \rightarrow \ce{YCH_2-CH_2X}\]
Naturaleza del Agente Sustitutivo
Es importante darse cuenta de que en la sustitución aromática el agente sustitutivo electrófilo real,\(\overset{\oplus}{\ce{X}}\) o\(\overset{\delta \oplus}{\ce{X}}-\overset{\delta \ominus}{\ce{Y}}\), no es necesariamente el reactivo que se añade a la mezcla de reacción. Por ejemplo, la nitración en mezclas de ácidos nítrico y sulfúrico no se produce por el ataque de la molécula de ácido nítrico sobre el compuesto aromático, sino por el ataque de una especie más electrófila, el ion nitronio,\(\ce{NO_2^+}\). Este ion se forma a partir de ácido nítrico y ácido sulfúrico de acuerdo con la siguiente ecuación:
\[\ce{HNO_3} + 2 \ce{H_2SO_4} \rightleftharpoons \ce{NO_2^+} + \ce{H_3O^+} + 2 \ce{HSO_4^-}\]
El ion nitrónico ataca el anillo aromático para dar primero un ion nitrobencenio y luego un compuesto nitro aromático:
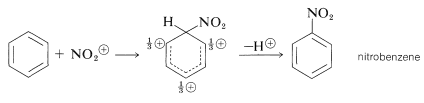
En general, la función de un catalizador (que a menudo es necesario para promover la sustitución aromática) es generar un agente sustitutivo electrófilo a partir de los reactivos dados. Por lo tanto, es necesario considerar cuidadosamente para cada reacción de sustitución cuál puede ser el agente sustitutivo real. Este problema no surge en el mismo grado en las adiciones electrofílicas a los alquenos, debido a que los alquenos son mucho más reactivos que los arenos que los reactivos empleados (por ejemplo\(\ce{Br_2}\),\(\ce{Cl_2}\),,\(\ce{HCl}\),\(\ce{HOCl}\),\(\ce{HOBr}\),\(\ce{H_3O}^\oplus\)) en sí mismos son suficientemente electrófilos para reaccionar con alquenos sin la ayuda de un catalizador. De hecho, las condiciones que conducen a la sustitución de arenos, como la nitración en mezclas de ácido nítrico y sulfúrico, a menudo degradarán el esqueleto de carbono de los alquenos.
Ahora consideraremos las reacciones de sustitución individuales enumeradas en la Figura 22-1 con respecto a la naturaleza del agente sustitutivo y la utilidad para la síntesis de diversas clases de compuestos aromáticos.
Nitración
El ion nitronio,\(\ce{NO_2^+}\), es el agente nitrante activo en mezclas de ácido nítrico-ácido sulfúrico. La nitración del metilbenceno (tolueno) es un ejemplo típico de una nitración que procede bien usando ácido nítrico en una mezcla 1:2 con ácido sulfúrico. El producto de nitración es una mezcla de 2-, 3- y 4-nitrometilbencenos:
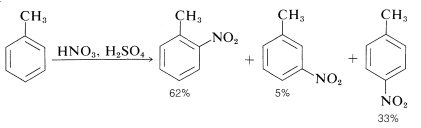
La presencia de cantidades apreciables de agua en la mezcla de reacción es perjudicial debido a que el agua tiende a revertir la reacción por la que se forma el ion nitrónico:
\[\ce{NO_2^+} + \ce{H_2O} \overset{\ce{HSO_4^-}}{\rightleftharpoons} \ce{HNO_3} + \ce{H_2SO_4}\]
Por esta razón, la potencia de una mezcla de ácido nítrico-sulfúrico puede aumentarse considerablemente mediante el uso de ácidos nítrico fumante y sulfúrico fumante. Con tales mezclas se puede lograr la nitración de compuestos relativamente no reactivos. Por ejemplo, el 4-nitrometilbenceno es mucho menos reactivo que el metilbenceno, pero cuando se calienta con un exceso de ácido nítrico en ácido sulfúrico fumante, se puede convertir sucesivamente en 2,4-dinitrometilbenceno y en 2,4,6-trinitrometilbenceno (TNT):
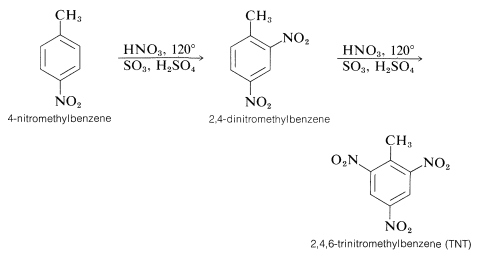
Hay varias características interesantes sobre las reacciones de nitración discutidas hasta ahora. Por ejemplo, las condiciones requeridas para la nitración del 4-nitrobetilbenceno oxidarían rápidamente un alqueno por escisión del doble enlace:
También la mononitración del metilbenceno no conduce a cantidades iguales de los tres productos posibles. El sustituyente metilo aparentemente orienta el sustituyente entrante preferentemente a las posiciones 2 y 4. Este aspecto de la sustitución aromática se discutirá en la Sección 22-5 junto con el efecto de los sustituyentes sobre la reactividad de los compuestos aromáticos.
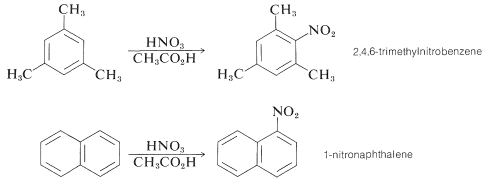
Algunos compuestos son suficientemente reactivos para que puedan nitrarse con ácido nítrico en ácido etanoico. Ejemplos pertinentes son 1,3,5-trimetilbenceno y naftaleno:
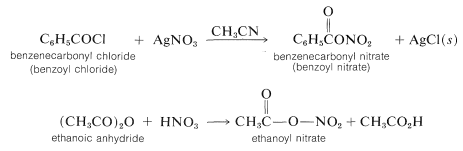
Otros reactivos de nitración convenientes son el nitrato de benzoilo y el nitrato de etanoilo\(\ce{CH_3COONO_2}\).\(\ce{C_6H_5COONO_2}\) Estos reactivos proporcionan una fuente de\(\ce{NO_2^+}\) y tienen alguna ventaja sobre\(\ce{HNO_3} \cdot \ce{H_2SO_4}\) las mezclas, ya que son solubles en disolventes orgánicos como etanitrilo o nitrometano. Tener soluciones homogéneas es especialmente importante para los estudios cinéticos de nitración. Los reactivos generalmente se preparan en solución según se requiera a partir del cloruro de acilo y nitrato de plata correspondientes o del anhídrido de ácido y ácido nítrico. Dichos reactivos son materiales peligrosos y deben ser manejados con cuidado.
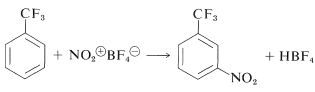
Las sales de nitronio del tipo\(\ce{NO_2^+} \ce{X^-}\) son agentes nitrantes muy potentes. El contraión,\(\ce{X^-}\), debe ser no nucleofílico y generalmente es fluoroborato,\(\ce{BF_4^-}\) o\(\ce{SbF_4^-}\):
Halogenación
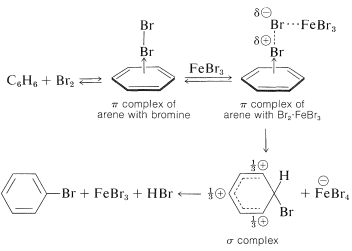
Hasta cierto punto hemos simplificado sobre-simplificamos la sustitución electrófila al descuidar el posible papel de los complejos 1:1 de transferencia de carga que la mayoría de los electrófilos forman con arenos (ver Sección 10-3C para la discusión de complejos análogos de alquenos):
Con halógenos, especialmente yodo, la formación de complejos es visualmente evidente a partir del color de las soluciones del halógeno en arenos. Aunque la formación de complejos puede ayudar a la sustitución al poner el halógeno y el areno en estrecha proximidad, la sustitución no ocurre necesariamente. Normalmente se requiere un catalizador, y los catalizadores más frecuentemente utilizados son haluros metálicos que son capaces de aceptar electrones (es decir, ácidos de Lewis tales como\(\ce{FeBr_3}\)\(\ce{AlCl_3}\), y\(\ce{ZnCl_2}\)). Su actividad catalítica puede atribuirse a su capacidad para polarizar el enlace halógeno-halógeno de la siguiente manera:
\[\overset{\delta \oplus}{\ce{Br}} \cdots \overset{\delta \ominus}{\ce{Br}} \cdots \ce{FeBr_3}\]
El extremo positivo del dipolo ataca al compuesto aromático mientras que el extremo negativo está complejado con el catalizador. Podemos representar la secuencia de reacción de la siguiente manera, siendo la etapa lenta la formación de un\(\sigma\) enlace entre\(\ce{Br}^\oplus\) y el anillo aromático:
El orden de reactividad de los halógenos es\(\ce{F_2} > \ce{Cl_2} > \ce{Br_2} > \ce{I_2}\). El flúor es demasiado reactivo para ser de uso práctico para la preparación de compuestos aromáticos de flúor y son necesarios métodos indirectos (ver Sección 23-10B). El yodo generalmente no es reactivo. Se ha afirmado que la yodación falla debido a que la reacción se invierte como resultado de las propiedades reductoras del yoduro de hidrógeno que se forma:
\[\ce{C_6H_6} + \ce{I_2} \overset{\rightarrow}{\longleftarrow} \ce{C_6H_5I} + \ce{HI}\]
Esta visión no es correcta porque, como demostró el propio Kekule, el yodobenceno no es reducido por el ácido yodhídrico excepto a temperaturas bastante altas.
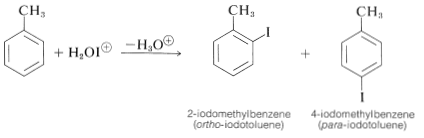
La manera de lograr la yodación directa en ausencia de potentes grupos sustituyentes activadores es convertir el yodo molecular en algunas especies más activas (quizás\(\ce{H_2OI}^\oplus\) o\(\ce{I}^\oplus\)) con un agente oxidante como el ácido nítrico o el peróxido de hidrógeno:
\[\begin{align} \ce{I_2} + 4 \ce{HNO_3} &\rightarrow 2 \ce{H_2O-I^+} + 2 \ce{NO_2} + 2 \ce{NO_3^-} \\ \ce{I_2} + \ce{H_2O_2} + 2 \ce{H^+} &\rightarrow 2 \ce{H_2OI^+} \end{align}\]
Con combinaciones de este tipo se obtienen buenos rendimientos de productos de yodación:
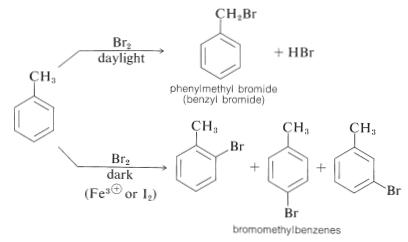
Las reacciones de sustitución de halógenos con cloro o bromo deben llevarse a cabo con una protección adecuada contra la luz fuerte. Si no se toman tales precauciones, un alquilbenceno reaccionará rápidamente con halógeno mediante un proceso fotoquímico para sustituir un hidrógeno del grupo alquilo en lugar del anillo aromático. La reacción tiene un mecanismo de cadena radical inducido por la luz del tipo discutido para la cloración del propeno (Sección 14-3A). Así, el metilbenceno reacciona con el bromo cuando se ilumina para dar bromuro de fenilmetilo; pero cuando se excluye la luz y está presente un catalizador ácido de Lewis, se produce la sustitución para dar principalmente los 2- y 4-bromometilbencenos. Se forma mucho menos del 3-bromometilbenceno:
El propio benceno puede inducirse a añadir halógenos en irradiación fuerte para dar polihalociclohexanos (ver Secciones 21-3A y 22-9C):
Alquilación
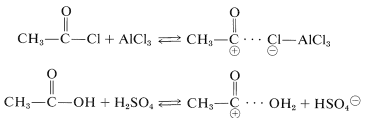
Un método importante de síntesis de alquilbencenos utiliza un haluro de alquilo como agente alquilante y un haluro metálico, generalmente cloruro de aluminio, como catalizador:
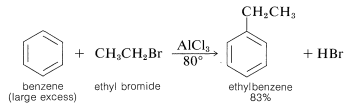
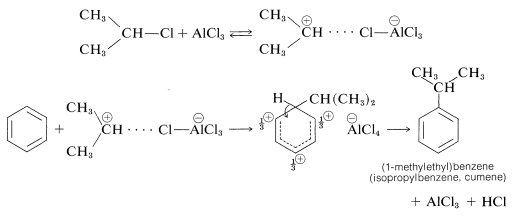
Esta clase de reacción se llama alquilación Friedel-Crafts en honor a sus descubridores, C. Friedel (un químico francés) y J. M. Crafts (un químico estadounidense). El catalizador de haluro metálico funciona tanto como lo hace en las reacciones de halogenación para proporcionar una fuente de un agente de sustitución positivo, que en este caso es un carbocatión:
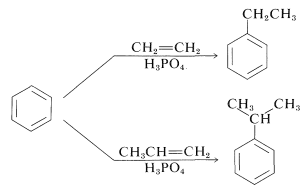
La alquilación no se limita a haluros de alquilo; se pueden usar alcoholes y alquenos de manera ventajosa en presencia de catalizadores ácidos tales como\(\ce{H_3PO_4}\),\(\ce{H_2SO_4}\),\(\ce{HF}\),\(\ce{BF_3}\), o\(\ce{HF-BF_3}\). El etilbenceno se elabora comercialmente a partir de benceno y eteno usando ácido fosfórico como catalizador. El isopropilbenceno se fabrica de manera similar a partir de benceno y propeno:
Bajo estas condiciones el carbocatión, que es el agente sustitutivo activo, se genera por protonación del alqueno:
\[\begin{align} \ce{CH_2=CH_2} + \ce{H^+} &\rightleftharpoons \ce{CH_3CH_2^+} \\ \ce{CH_3CH=CH_2} + \ce{H^+} &\rightleftharpoons \ce{CH_3} \overset{+}{\ce{C}} \ce{HCH_3} \end{align}\]
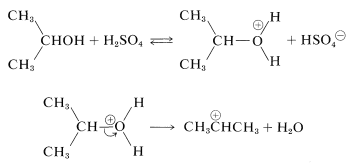
Con alcoholes el electrófilo se puede formar por protonación inicial por el catalizador ácido y posterior escisión a un carbocatión:
Limitaciones de las reacciones de alquilación
Polisustitución
Existen varios factores que limitan la utilidad de las reacciones de alquilación. Primero, puede ser difícil limitar la reacción a la monosustitución porque la introducción de un sustituyente alquilo tiende a activar el anillo hacia una segunda sustitución (ver Sección 22-5). Por lo tanto, para obtener rendimientos razonables de un monoalquilbenceno, generalmente es necesario usar un gran exceso en relación con el agente alquilante:
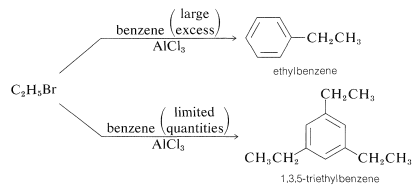
Reordenamiento del agente alquilante
Una segunda limitación es la inclinación por el reactivo alquilante para dar productos de reordenamiento. Como ejemplo, la alquilación de benceno con 1-cloropropano conduce a una mezcla de propilbenceno e isopropilbenceno. Podemos escribir la reacción como primero implicando la formación de un catión propilo, que es un carbocatión primario:
\[\ce{CH_3CH_2CH_2Cl} + \ce{AlCl_3} \rightarrow \ce{CH_3CH_2CH_2^+} + \overset{-}{\ce{Al}} \ce{Cl_4}\]
Este ion puede alquilar benceno para dar propilbenceno,
\[\ce{C_6H_6} + \ce{CH_3CH_2CH_2^+} \rightarrow \ce{C_6H_5CH_2CH_2CH_3} + \ce{H^+}\]
o puede reorganizarse a un ion secundario más estable mediante la transferencia de un hidrógeno de un carbono vecino junto con su par de electrones de enlace (es decir, desplazamiento de 1,2-hidruro). La carga positiva se transfiere de\(\ce{C_1}\) a\(\ce{C_2}\):
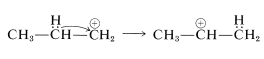
La alquilación de benceno con el catión isopropilo produce entonces isopropilbenceno:
\[\ce{C_6H_6} + \ce{CH_3} \overset{\oplus}{\ce{C}} \ce{HCH_3} \rightarrow \ce{C_6H_5CH(CH_3)_2} + \ce{H}^\oplus\]
Los reordenamientos de este tipo que involucran intermedios de carbocationes a menudo ocurren en alquilaciones de Friedel-Crafts con grupos alquilo primarios y secundarios mayores que\(\ce{C_2}\) y\(\ce{C_3}\). Los reordenamientos de carbocationes relacionados se analizan en las Secciones 8-9B y 15-5E.
Reordenamiento de productos
Otras complicaciones surgen del hecho de que las reacciones de alquilación a veces están bajo control de equilibrio en lugar de control cinético. Los productos a menudo isomerizan y son desproporcionados, particularmente en presencia de grandes cantidades de catalizador. Así, 1,2- y 1,4-dimetilbencenos (orto - y para -xilenos) se convierten por grandes cantidades de catalizadores Friedel-Crafts en 1,3-dimetilbenceno (meta - xileno):
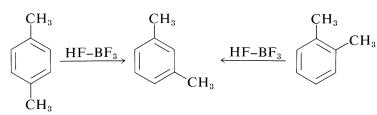
El etilbenceno se desproporciona bajo la influencia del exceso\(\ce{HF-BF_3}\) a benceno y 1,3-dietilbenceno:
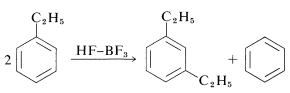
Acilación
La acilación y alquilación de arenos están estrechamente relacionadas. Ambas reacciones se desarrollaron como resultado de la colaboración entre Friedel y Crafts, en 1877. La reacción de acilación introduce un grupo acilo\(\ce{RCO}\), en un anillo aromático y el producto es una arilcetona:
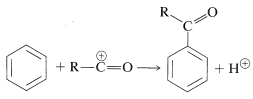
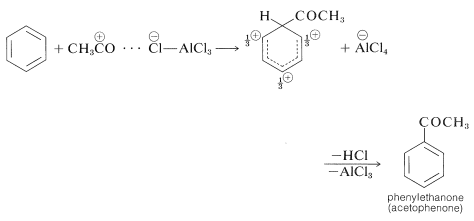
Los reactivos acilantes comúnmente usados son haluros de ácido carboxílico\(\ce{RCOCl}\),, anhidridos\(\ce{(RCO)_2O}\), o el ácido mismo\(\ce{RCO_2H}\). Un protón fuerte u otro catalizador ácido de Lewis es esencial. El catalizador funciona para generar el catión acilo:
El catalizador más comúnmente utilizado con haluros y anhidridos de acilo es el cloruro de aluminio:
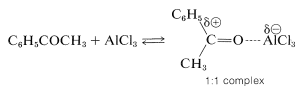
La acilación difiere de la alquilación en que la reacción generalmente se lleva a cabo en un disolvente, comúnmente disulfuro de carbono\(\ce{CS_2}\), o nitrobenceno. Además, la acilación requiere más catalizador que alquilación, debido a que gran parte del catalizador está atado e inactivado por la formación de complejos con el producto cetona:
A diferencia de la alquilación, la acilación se controla fácilmente para dar monosustitución, ya que una vez que un grupo acilo está unido a un anillo de benceno, no es posible introducir un segundo grupo acilo en el mismo anillo. Debido a esto, una síntesis conveniente de alquilbencenos comienza con la acilación, seguida de la reducción del grupo carbonilo con zinc y ácido clorhídrico (Sección 16-6). Por ejemplo, el propilbenceno se prepara mejor por esta ruta de dos etapas porque, como hemos señalado, la alquilación directa de benceno con cloruro de propilo produce cantidades considerables de isopropilbenceno y productos de polisustitución:
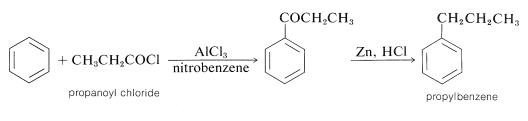
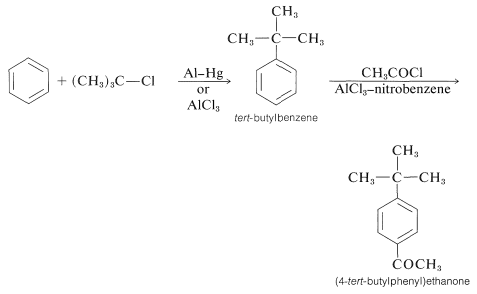
En la acilación del alquilbenceno el producto casi siempre es el isómero para. La síntesis de (4-terc- butilfenil) etanona ilustra esto así como el uso secuencial de reacciones de alquilación y acilación:
Los químicos se inclinan a dar nombres a reacciones que los asocian ya sea con sus descubridores o con los productos que dan. Esta práctica puede resultar confusa porque muchas reacciones nombradas (o “reacciones de nombre”) que alguna vez se pensó que no tenían ninguna relación, han resultado tener mecanismos muy similares. Así tenemos dos reacciones de acilación estrechamente relacionadas: una es la síntesis de cetona de Friedel-Crafts, en la que está el electrófilo\(\ce{R} \ce{-} \overset{\oplus}{\ce{C}} \ce{=O}\); y la otra es la síntesis de aldehído Gattermann-Koch, en la que el electrófilo es\(\ce{H} \ce{-} \overset{\oplus}{\ce{C}} \ce{=O}\):
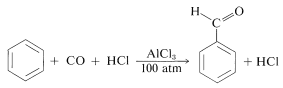
Esta última reacción utiliza monóxido de carbono y\(\ce{HCl}\) bajo presión en presencia de cloruro de aluminio. Se puede considerar que el electrófilo está formado de la siguiente manera:
\[\ce{C=O} + \ce{HCl} + \ce{AlCl_3} \rightleftharpoons \ce{H} \ce{-} \overset{\oplus}{\ce{C}} \ce{=O} + \overset{\ominus}{\ce{Al}} \ce{Cl_4}\]
Sulforación
La sustitución del\(\left( \ce{-SO_3H} \right)\) grupo ácido sulfónico por un hidrógeno de un hidrocarburo aromático se puede llevar a cabo calentando el hidrocarburo con un ligero exceso de ácido sulfúrico concentrado o fumante:
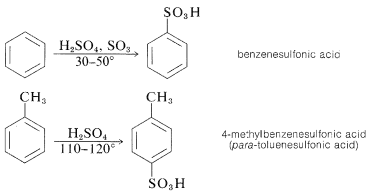
El agente sulfonante real normalmente es la\(\ce{SO_3}\) molécula, que, aunque neutra, tiene un átomo de azufre poderosamente electrófilo:
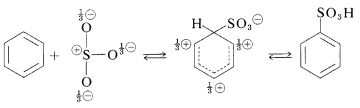
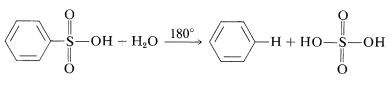
La sulfonación es reversible y el\(\ce{-SO_3H}\) grupo se puede eliminar por hidrólisis en\(180^\text{o}\):
Una preparación alternativa útil de derivados de sulfonilo es posible con ácido clorosulfónico:
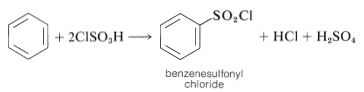
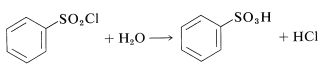
Este procedimiento tiene una ventaja sobre la sulfonación directa, ya que los cloruros de sulfonilo suelen ser solubles en disolventes orgánicos y pueden separarse fácilmente de la mezcla de reacción. Además, el cloruro de sulfonilo es un intermedio más útil que el ácido sulfónico, pero puede convertirse en el ácido por hidrólisis si se desea:
La sulfonación es importante en la producción comercial de una clase importante de detergentes - los alquilbencenosulfonatos de sodio:
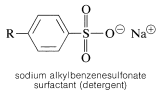
La síntesis ilustra varios tipos importantes de reacciones que hemos discutido en este y en capítulos anteriores. En primer lugar, el grupo alquilo\(\ce{R}\) suele ser un\(\ce{C_{12}}\) grupo derivado del hidrocarburo de cadena lineal, dodecano, que en la fotocloración da una mezcla de clorododecanos:
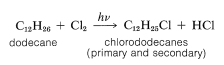
Esta mezcla de clorododecanos se emplea para alquilar benceno, dando así una mezcla de dodecilbencenos isoméricos, llamada alquilato detergente:
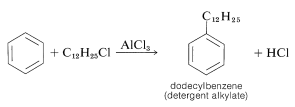
La sulfonación del alquilato detergente da exclusivamente los ácidos 4-dodecilbencensulfónicos, que con hidróxido de sodio forman dodecilbencenosulfonatos solubles en agua:
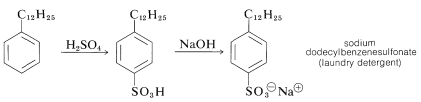
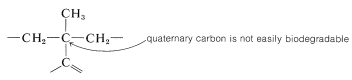
En muchos países está prohibido por ley comercializar detergentes de este tipo, que tienen grupos alquilo altamente ramificados. La razón es que los carbonos cuaternarios y, en menor medida, los carbonos terciarios no son degradados fácilmente por las bacterias en las plantas de tratamiento de aguas residuales:
Intercambio de Hidrógeno
Es posible reemplazar los hidrógenos del anillo de muchos compuestos aromáticos por intercambio con ácidos fuertes. Cuando se usa un ácido isotópicamente marcado como\(\ce{D_2SO_4}\) el mismo, esta reacción es una manera fácil de introducir deuterio. El mecanismo es análogo a otras sustituciones electrofílicas:
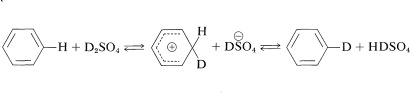
El perdeuteriobenceno se\(^3\) puede elaborar a partir de benceno con buen rendimiento si se utiliza un exceso suficientemente grande de ácido deuteriosulfurico. La deuteración puede parecer competitiva con la sulfonación, pero la deuteración ocurre en condiciones mucho más suaves.
Sustitución aromática por metalación electrofílica
Debido a que los metales son elementos electropositivos, pueden considerarse electrófilos potenciales. Sus reacciones con arenos han sido investigadas más a fondo para detectar mercurio. El benceno puede sustituirse con\(\ce{HgX}^\oplus\) derivado de una sal mercúrica\(\ce{HgX_2}\), en presencia de un catalizador ácido. La sal más utilizada es el etanoato mercúrico,\(\ce{Hg(OOCCH_3)_2}\). Se considera que el catalizador funciona ayudando a la generación del electrófilo activo,\(\ce{HgX}^\oplus\). Otros metales que pueden introducirse directamente en un anillo aromático de esta manera incluyen el talio y el plomo.
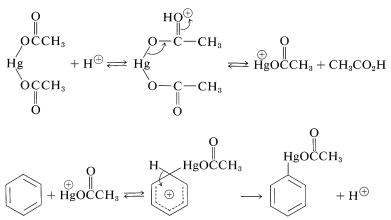
\(^3\)El prefijo per, como en perdeuterio- o perfluoro-, significa que todos los hidrógenos han sido reemplazados por el sustituyente nombrado,\(\ce{D}\) o\(\ce{F}\). Perhidro significa saturado o completamente hidrogenado.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."