
26.1: Compuestos de Arilo Oxígeno
- Page ID
- 73698

Fenoles (Arenoles) - Propiedades Físicas
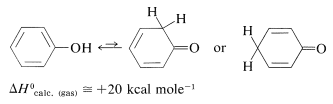
Los fenoles son enoles, y los enoles normalmente son inestables con respecto a los compuestos carbonílicos correspondientes (Sección 17-1D). Por lo tanto
La situación es diferente para los fenoles debido a la inclusión del doble enlace carbono-carbono en el anillo aromático y la estabilización aromática asociada. El fenol (bencenol) existe exclusivamente en la forma enol:
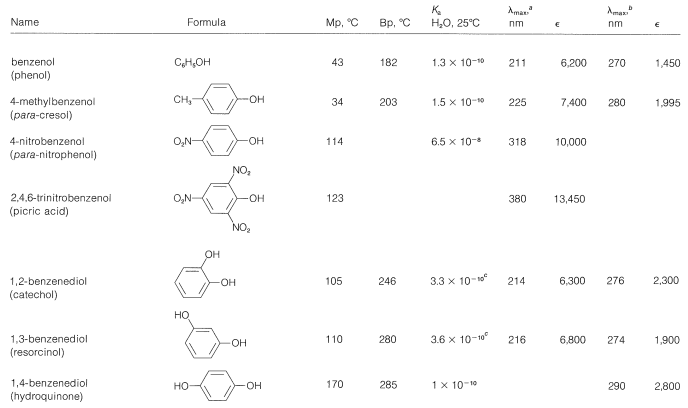
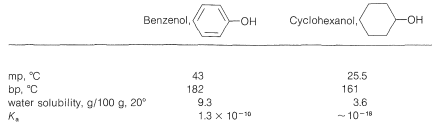
Las propiedades físicas de algunos fenoles representativos se resumen en el Cuadro 26-1. En general, los fenoles son algo más polares que los correspondientes alcoholes saturados. Las magnitudes de las diferencias se ilustran bien mediante la comparación de las propiedades físicas del bencenol y el ciclohexanol, que se muestran en el Cuadro 26-2. El factor determinante parece ser la mayor acidez del grupo hidroxilo fenólico, lo que significa que, en la forma no disociada, el\(\ce{O-H}\) enlace está más fuertemente polarizado\(\overset{\delta \ominus}{\ce{O}}-\overset{\delta \oplus}{\ce{H}}\) que para los alcoholes. Por lo tanto, los fenoles forman enlaces de hidrógeno más fuertes que los alcoholes, lo que resulta en puntos de ebullición más altos, mayor solubilidad en agua y mayor capacidad para actuar como solventes para moléculas orgánicas razonablemente polares.
Cuadro 26-1: Propiedades físicas de algunos fenoles representativos
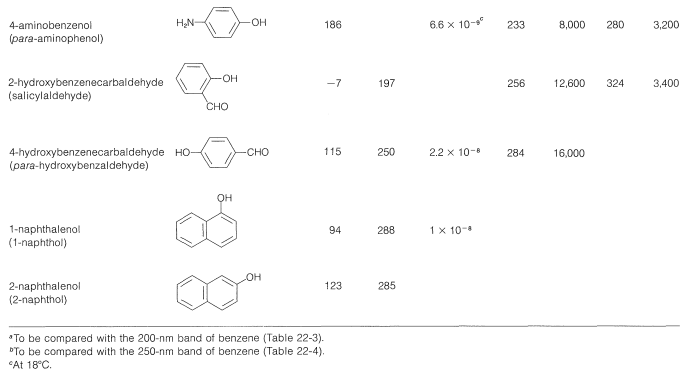
Las longitudes de onda de los máximos de absorción ultravioleta de los arenoles mostrados en el Cuadro 26-1 indican un efecto considerable de los sustituyentes sobre estas absorciones, que corresponden a las\(\text{nm}\) absorciones\(200\)\(255\) -\(\text{nm}\) y - del benceno (Sección 22-3B).
Cuadro 26-2: Propiedades Físicas Comparativas de Bencenol y Ciclohexanol
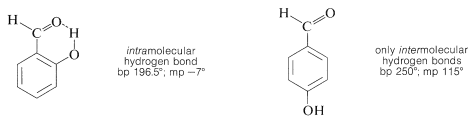
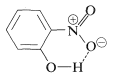
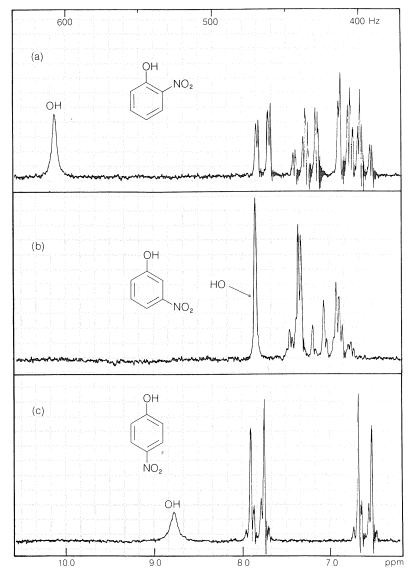
Sustancias como el 2-hidroxibenzaldehído, el ácido 2-hidroxibenzoico y el 2-nitrobencenol forman enlaces de hidrógeno intra- en lugar de intermoleculares. Esto reduce efectivamente la atracción intermolecular, reduciendo así los puntos de ebullición y aumentando la solubilidad en disolventes no polares en comparación con los isómeros meta y para, que solo forman enlaces de hidrógeno intermoleculares:
Síntesis de fenoles
El benzenol y los 2-, 3- y 4-metilbencenoles (cresoles) pueden aislarse del alquitrán de hulla (Sección 22-11). El propio benzenol se usa comercialmente en cantidades tan grandes que son necesarios métodos alternativos de preparación y la mayoría de estos comienzan con benceno o alquilbencenos. La oxidación directa del benceno no es satisfactoria porque el bencenol se oxida más fácilmente que el benceno.
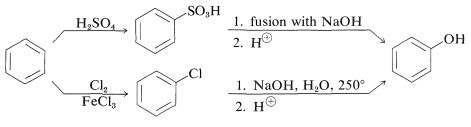
En un momento, el bencenol se hizo industrialmente sulfonando o clorando benceno y luego introduciendo el grupo hidroxilo por sustitución nucleófila con álcali fuerte:
Las síntesis comerciales actuales del bencenol implican la oxidación de metilbenceno o isopropilbenceno (Sección 16-9E). La oxidación de isopropilbenceno es económicamente factible para la producción de bencenol porque la 2-propanona (acetona) también es un producto valioso:
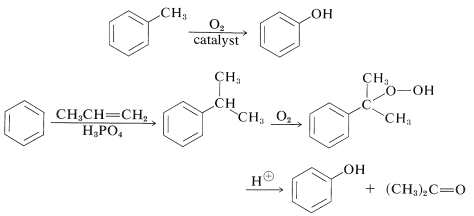
Un procedimiento común de laboratorio convierte una amina aromática en un fenol por medio de la sal de arenodiazonio,\(\ce{ArNH_2} \rightarrow \ce{ArN_2^+} \rightarrow \ce{ArOH}\) (Sección 23-10B).
Reacciones de fenoles que involucran los\(\ce{O-H}\) enlaces
Las reacciones de los grupos hidroxilo de los fenoles, en donde los\(\ce{O-H}\) enlaces están rotos, son similares a las de los alcoholes. Así, los fenoles son ácidos débiles (\(K_a = 10^{-10}\)a\(10^{-8}\); Cuadro 26-1), intermedios en fuerza entre ácidos carboxílicos y alcoholes.
Los enoles son ácidos más fuertes que los alcoholes debido al aumento en la deslocalización de electrones en aniones enolatos en comparación con los enoles neutros (ver Sección 15-8A). La energía de estabilización del bencenol (Cuadro 21-1) es\(48 \: \text{kcal mol}^{-1}\),\(5 \: \text{kcal}\) mayor que la del benceno. Podemos atribuir este aumento a la deslocalización de un par de electrones no compartidos del oxígeno:
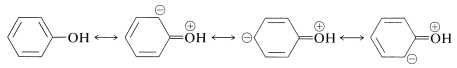
Cuando el\(\ce{OH}\) protón es eliminado por una base, el anión resultante tiene una estabilización aún mayor, ya que las estructuras de enlace de valencia correspondientes no implican separación de carga:
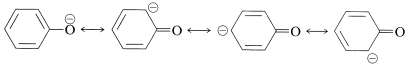
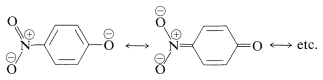
Podemos estar seguros de que los grupos sustituyentes que estabilizan el anión aumentarán la acidez. Así, el 4-nitrobencenol es aproximadamente 500 veces más fuerte como ácido que el bencenol, debido a la mayor deslocalización de la carga al grupo nitro:
Es posible preparar ésteres de fenoles con anhídrido de ácido carboxílico o haluros de ácido, y éteres de fenilo por reacción del anión bencenolato con haluros, ésteres de sulfato, sulfonatos u otros derivados de alquilo que reaccionen bien por el\(S_\text{N}2\) mecanismo:
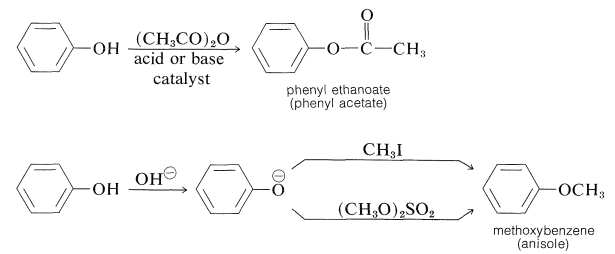
Los fenoles (como los ácidos carboxílicos; Sección 24-7C y Tabla 18-6) se convierten en derivados metoxi con diazometano:
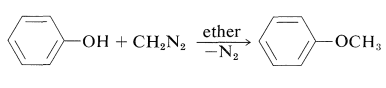
Casi todos los fenoles y enoles (como los de 1,3-dicetonas) dan colores con cloruro férrico en agua diluida o soluciones de alcohol. El propio benzenol produce un color violeta con cloruro férrico, y los metilbencenoles dan un color azul. Los productos aparentemente son sales de arenolato férrico, que absorben la luz visible para dar un estado excitado teniendo electrones deslocalizados tanto sobre el átomo de hierro como sobre el sistema insaturado.
Reacciones de fenoles que involucran los\(\ce{C-O}\) enlaces
En general, es muy difícil romper el\(\ce{C-O}\) enlace aromático de los arenoles. Así, los ácidos halogenados concentrados no convierten arenoles simples en haluros de arilo, y los alcoxiarenos se escinden con bromuro de hidrógeno o yoduro de hidrógeno de la manera en\(\ce{ArO-R}\) lugar de\(\ce{Ar-OR}\):
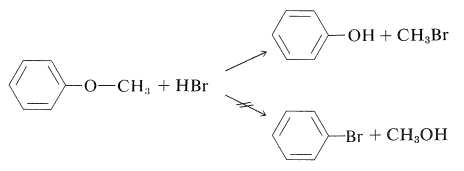
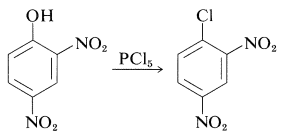
(Los éteres de diarilo, como el éter de difenilo, no reaccionan con yoduro de hidrógeno incluso a\(200^\text{o}\).) No hay una manera fácil de convertir arenoles en haluros de arilo, excepto cuando la activación es proporcionada por grupos 2- o 4-nitro. Así, el 2,4-dinitrobencenol se convierte en 1-cloro-2,4-dinitrobenceno con pentacloruro de fósforo:
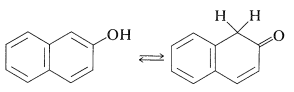
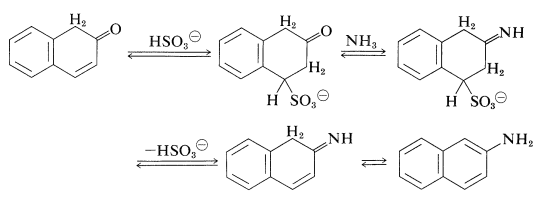
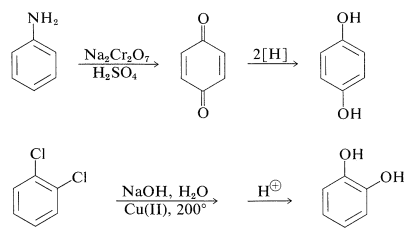
Una excepción a la generalización de que\(\ce{C-O}\) los enlaces a sistemas aromáticos son difíciles tanto de hacer como de romper es proporcionada por la conversión reversible de bencenodioles y 1- o 2-naftalenoles a las aminas correspondientes, generalmente a temperaturas elevadas con hidrogenosulfito de sodio o un catalizador ácido. La reacción inducida por sulfito de hidrógeno de sodio se llama reacción de Bucherer:
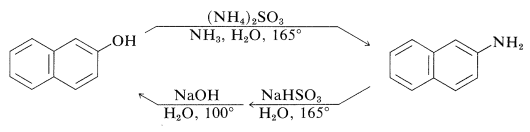
Estas reacciones no funcionan bien con los bencenoles simples porque el paso clave es la formación del isómero cetodel arenol, un proceso que es desfavorable para los bencenoles simples.
 \(\tag{26-1}\)
\(\tag{26-1}\)
El papel del sulfito de hidrógeno es la participación en una adición 1,4-reversible a la cetona insaturada para retenerla en forma de cetona que luego se convierte en la imina por\(\ce{NH_3}\) (Sección 16-4C) y por lo tanto a la arenamina:
Reacciones de sustitución en los carbonos del anillo de arenoles
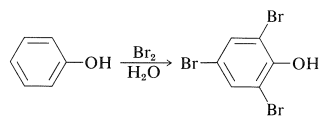
Los sistemas\(\pi\) orbitales ricos en electrones de los arenoles y especialmente de los iones arenolato hacen que estos compuestos sean muy susceptibles a la sustitución electrófila. Los arenoles típicamente reaccionan rápidamente con bromo en solución acuosa para sustituir las posiciones orto o para con respecto al grupo hidroxilo. El propio benzenol da 2,4,6-tribromobenzenol con alto rendimiento:
Varias reacciones importantes de arenoles implican la sustitución aromática de iones arenolato con electrófilos de carbono. En cierto sentido, estas reacciones son reacciones de alquilación y acilación como se discute para los arenos (Secciones 22-4E y 22-4F). En otro sentido, son reacciones de alquilación y acilación de aniones enolatos y por lo tanto podrían dar lugar a productos por\(\ce{C}\) - y\(\ce{O}\) -alquilación, o\(\ce{C}\) - y\(\ce{O}\) -acilación (Sección 17-4). Por lo tanto:
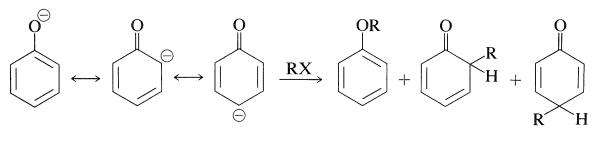
En la mayoría de los casos, predomina\(\ce{O}\) la -alquilación. Sin embargo, con haluros de 2-propenilo, cualquiera de las reacciones se puede hacer esencialmente la reacción exclusiva mediante la elección apropiada del disolvente. Con el bencenolato de sodio los disolventes más polares, como la 2-propanona, conducen al 2-propeniloxibenceno, mientras que en los disolventes no polares, como el benceno, el 2- (2-propenil) bencenol es el producto favorecido:
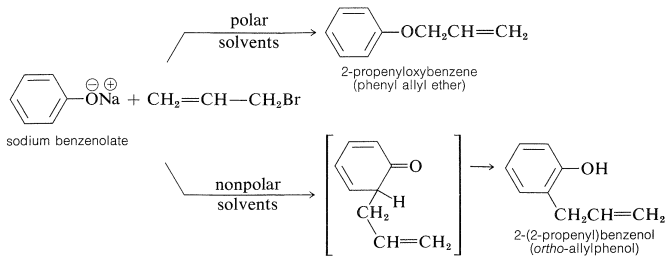
Cabe señalar que la formación de 2- (2-propenil) bencenol en disolventes no polares no es el resultado de la\(\ce{O}\) -propenilación seguida de reordenamiento, aunque el producto de\(\ce{C}\) -propenilación es termodinámicamente más estable. De hecho, el reordenamiento ocurre, pero a temperaturas mucho más altas (superiores\(200^\text{o}\)) que las requeridas para propenilar el bencenolato de sodio:
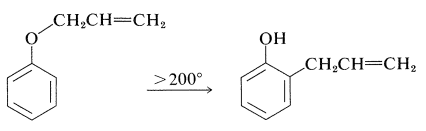
Tales reordenamientos son bastante generales para aril alil-éteres y se denominan reordenamientos de Claisen. Son ejemplos de las reacciones pericílicas discutidas en la Sección 21-10D.
La reacción de Kolbe-Schmitt produce\(\ce{O}\) - y\(\ce{C}\) -carboxilación a través de la reacción de dióxido de carbono con bencenolato de sodio en\(125^\text{o}\):

El bencenolato de sodio absorbe dióxido de carbono a temperatura ambiente para formar fenil carbonato de sodio (\(\ce{O}\)-carboxilación) y, cuando éste se calienta a una\(125^\text{o}\) presión de varias atmósferas de dióxido de carbono, se reorganiza a 2-hidroxibenzoato de sodio (salicilato de sodio). Sin embargo, no hay evidencia de que esta reacción sea otra que un proceso de disociación-recombinación, en el que el paso importante implica el ataque electrófilo por dióxido de carbono sobre el anillo aromático del ion bencenolato (\(\ce{C}\)-carboxilación):
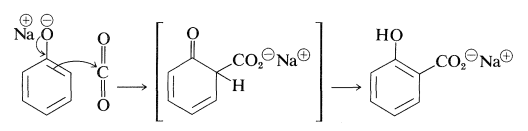
Con el bencenolato de sodio a temperaturas de\(125^\text{o}\) a\(150^\text{o}\), ocurre la sustitución orto; a temperaturas más altas (\(250^\text{o}\)to\(300^\text{o}\)), particularmente con la sal de potasio, se favorece el isómero para.
La reacción de Kolbe-Schmitt se relaciona con carboxilaciones enzimáticas como de\(D\) -ribulosa-1,5-difosfato con dióxido de carbono, un paso clave en la fotosíntesis (Sección 20-9). El resultado global es la formación de\(\ce{C-C}\) enlaces por adición de\(\ce{CO_2}\) una sal enolato o su equivalente enamina.
En la reacción de Reimer-Tiemann algo relacionada, el bencenolato de sodio con triclorometano en solución alcalina forma la sal sódica del 2-hidroxibencenocarbaldehído (salicilaldehído). El electrófilo en este caso probablemente sea diclorocarbeno (Sección 14-7B):
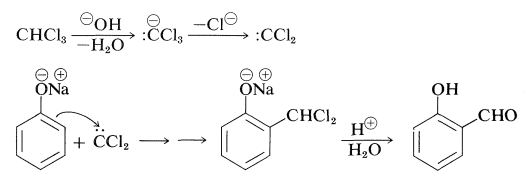
Muchos fenoles experimentan reacciones de adición de tipo aldol con compuestos carbonílicos en presencia de ácidos o bases. Así, el bencenol reacciona con metanal en condiciones alcalinas suaves para formar (4-hidroxifenil) metanol:
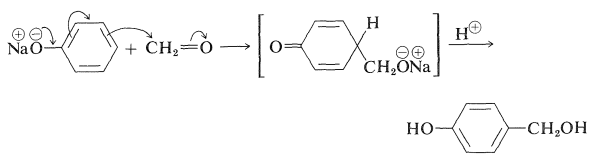
El uso de este tipo de reacción en la formación de polímeros se discutirá en el Capítulo 29.
Los arenoles suelen sufrir reacciones de acoplamiento diazo con sales de arildiazonio a valores de pH lo suficientemente altos como para convertir parte del arenol en los aniones arenolato nucleofílicos más potentes:
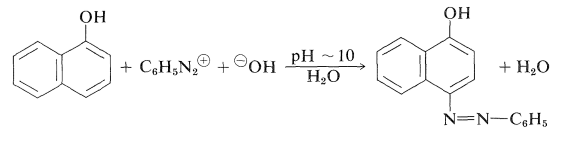
Sin embargo, si el pH es demasiado alto, se inhibe el acoplamiento debido a que se transforma en la sal de diazonio\(\ce{ArN=N-O}^\ominus\), que es la base conjugada no electrofílica de un ácido diazótico (Cuadro 23-4).
Reacciones de Adición
Los arenoles se pueden reducir exitosamente con catalizadores de hidrógeno sobre níquel a los ciclohexanoles correspondientes. Se puede preparar una variedad de ciclohexanoles alquil-sustituidos de esta manera:
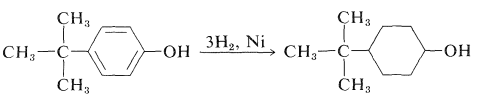
Oxidación de Arenoles
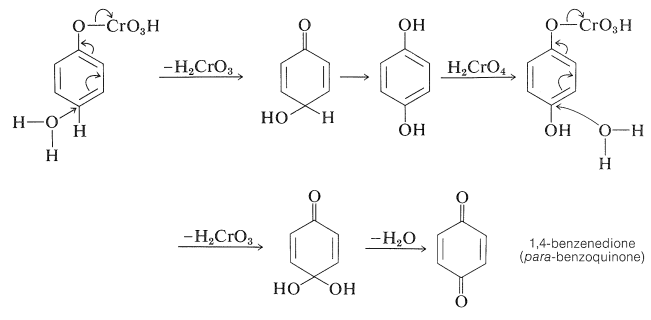
El benzenol se puede oxidar a 1,4-bencenodiona (para -benzoquinona) por el ácido crómico. La reacción puede proceder por medio de hidrógeno-cromato de fenilo (Sección 15-6B) como sigue:
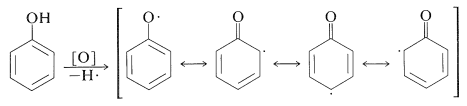
Las reacciones de oxidación de los arenoles con otros oxidantes son complejas. El ataque oxidativo parece implicar, como primer paso, la eliminación del hidrógeno hidroxilo para producir un radical fenoxi:
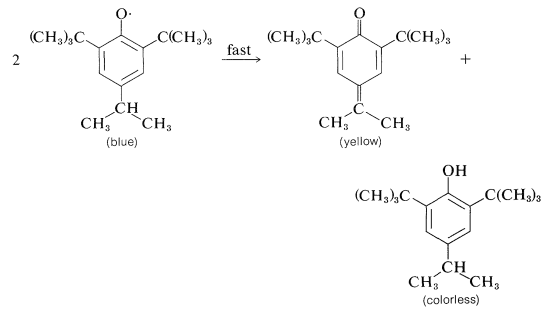
El curso posterior depende de los sustituyentes en el anillo aromático. Con 2,4,6-tri- terc- butilbencenol, el radical es razonablemente estable en solución de benceno y su presencia está indicada tanto por su color azul oscuro como por el hecho de que agrega al 1,3-butadieno:
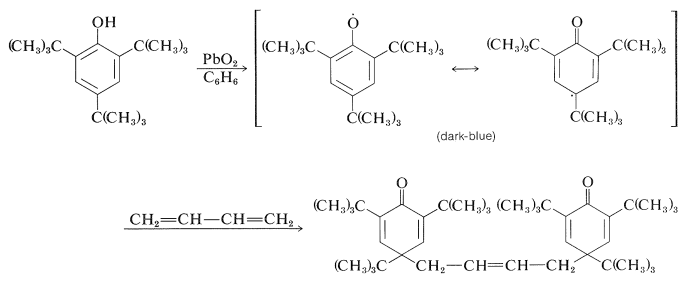
Aparentemente, la dimerización del radical fenoxi anterior a través del oxígeno o del anillo es inhibida por los voluminosos grupos terc- butilo. Con menos o menores sustituyentes, los radicales fenoxi pueden formar productos de dimerización o desproporción. A continuación se dan ejemplos de estas reacciones.
Dimerización:
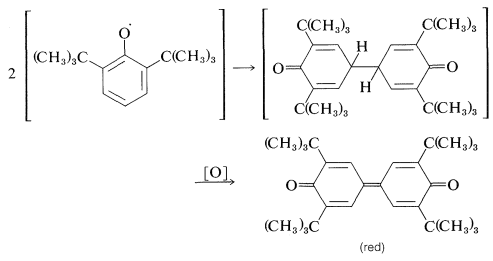
Desproporcionación:
Polioles de areno
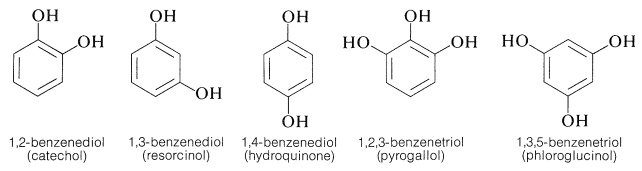
Varios compuestos aromáticos importantes tienen más de un grupo hidroxilo areno. Estos más a menudo son derivados de los siguientes arenoles dihídricos y trihídricos, todos los cuales tienen nombres de uso común (pero poco descriptivos):
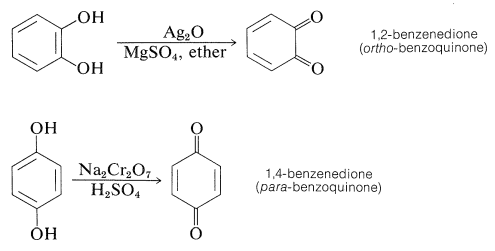
Todos son excepcionalmente reactivos con los reactivos electrofílicos, particularmente en solución alcalina, y todos se oxidan fácilmente. Los 1,2- y 1,4-bencenodioles, pero no el 1,3-bencenodiol, se oxidan a quinonas:
La preparación de estas sustancias se puede lograr mediante métodos estándar para sintetizar arenoles, pero la mayoría de ellas en realidad se realizan a escala comercial mediante procedimientos bastante especiales, algunos de los cuales se resumen de la siguiente manera:
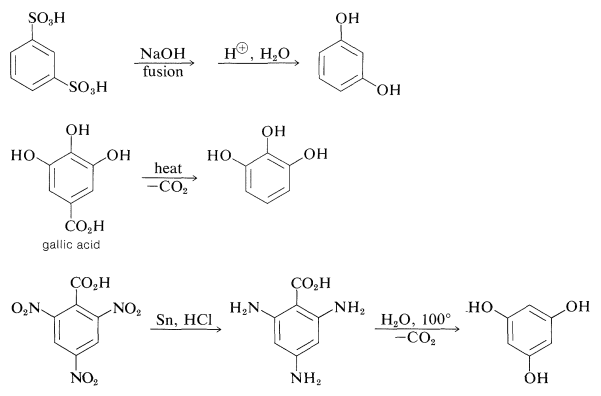
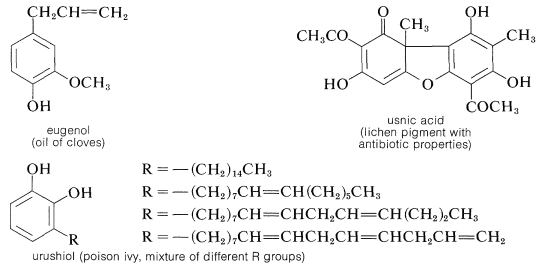
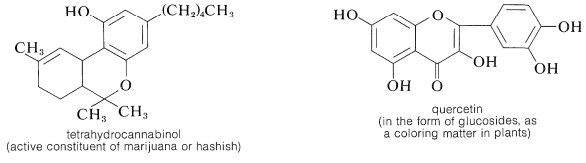
El ácido gálico utilizado en la preparación del 1,2,3-bencenotriol se puede obtener por degradación microbiana de taninos, los cuales son combinaciones complejas de glucosa y ácido gálico obtenidos de corteza de roble y gallnuts. Algunos otros representantes de los muchos tipos de derivados naturales de arenoles polihídricos son
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."