
1.27: Adiciones electrofílicas
- Page ID
- 74314

En el grupo funcional de un alqueno, el doble enlace carbono-carbono, los electrones más fácilmente disponibles son los del enlace pi. Están más lejos de los núcleos que los electrones en un enlace sigma, por lo que son atraídos más fácilmente a un electrófilo si uno se acerca. Otra forma de decir esto es que los electrones pi son débilmente nucleofílicos. Comenzaremos por observar algunas reacciones que comienzan con el ataque de un electrófilo en la parte pi del doble enlace.
Primero, repasemos brevemente una reacción del grupo carbonilo con un electrófilo. La adición catalizada por ácido de agua a un aldehído es una de esas reacciones discutidas anteriormente. El mecanismo es:
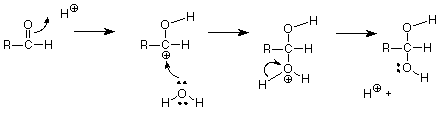
El primer paso es el ataque electrófilo sobre el enlace carbonilo pi por el ácido electrófilo H +. Este paso hace un carbocatión, que luego es atacado por el agua nucleófila débil pero muy abundante. (No use OH - como nucleófilo. Hay muy poco OH - ¡en una solución ácida! ) El paso final de “limpieza” da el producto y devuelve un H + para regenerar el catalizador.
Ahora vamos a aplicar este mismo mecanismo a la adición de agua al etileno, el alqueno más pequeño.
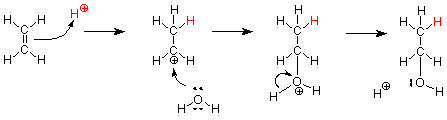
Observe que los pasos son los mismos y que los dos últimos pasos (lo que sucede con el carbocatión) son los mismos pasos que ocurrieron en la hidrólisis de S N 1 (solvolisis en agua) de un haluro de alquilo.
Estos pasos —primero un electrófilo ataca el enlace pi para formar un carbocatión, segundo un nucleófilo ataca al carbocatión— son los pasos clave en las reacciones más importantes de los alquenos, las reacciones de adición electrófila. El primer paso es el lento, por lo que es el que determina la velocidad de la reacción. Esto tiene consecuencias muy importantes cuando introducimos una ligera complicación en la estructura del alqueno.
Orientación - Regla de Markovnikov
¿Qué pasa si aplicamos esta reacción a un alqueno como el propeno? (Tal alqueno se llama “asimétrico” ya que los patrones de sustitución en los dos carbonos de alqueno son diferentes). Hay dos posibles productos ya que el OH podría terminar ya sea en el carbono CH 2 o en el carbono CH 3 CH.
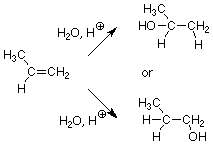
Podemos esperar razonablemente que el producto que se forma más rápido sea el que predomine en la mezcla de productos. De hecho, si un producto se forma más de 100 veces más rápido que el otro, es el único producto que observaremos en un sentido práctico. Esto significa que predecir qué producto se forma se reduce a predecir qué producto se forma más rápido. A su vez, esta pregunta se convierte en “¿qué producto está formado por una vía con una menor energía de activación?” Estimamos las energías de activación relativas observando las estructuras del estado de transición. La vía con el estado de transición de energía más baja será la vía más rápida. Producirá más producto en un tiempo dado que la vía más lenta, por lo que su producto se encontrará en la mayor cantidad.
¿Cómo hacemos tal predicción? Conocemos el mecanismo, por lo que podemos aplicarlo a las vías que conducirían a nuestros productos potenciales. Afortunadamente, las tasas asociadas a estas dos vías están determinadas por sus primeros pasos, por lo que solo necesitamos preocuparnos por las energías de activación para esos pasos.
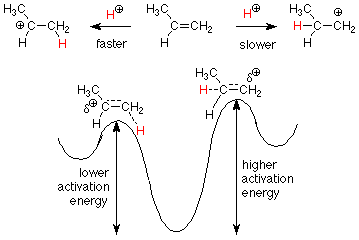
Sabemos que un carbocatión secundario (a la izquierda) es más estable (tiene una menor energía) que un carbocatión primario (a la derecha). Esto lo hemos explicado diciendo que los electrones en un grupo metilo son mejores para aliviar parcialmente la deficiencia de electrones del carbocatión que la relativa falta de electrones alrededor de un hidrógeno. Dado que los estados de transición para estos dos pasos también son carbocationes parcialmente formados deficientes en electrones, esperaríamos que prevalezcan los mismos efectos en los estados de transición. Esto nos dice que la reacción a la izquierda tendrá la menor energía de activación y ocurrirá más rápido. Después esperamos ver el producto en el que el H + atacante se une al carbono menos sustituido y el OH está unido al carbono más sustituido.
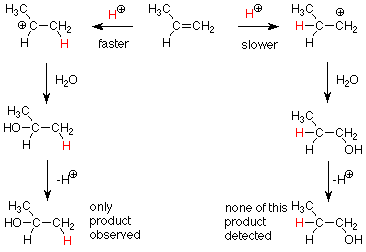
De manera más general, el electrófilo ataca al átomo de carbono menos sustituido en la primera etapa y el nucleófilo ataca al producto más sustituido en la segunda etapa.
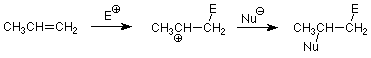
Podemos usar esto para predecir qué producto se formará en una adición electrófila a un alqueno asimétrico. Todo lo que tenemos que hacer es identificar el electrófilo y el nucleófilo en el compuesto que está agregando. Por ejemplo, considere la adición de HBr. El enlace HBr está polarizado de manera que el H es positivo y el Br es negativo. El H + es así el electrófilo y el Br - es el nucleófilo. La aplicación del patrón anterior nos da:
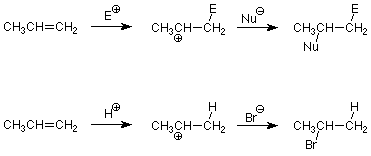
Históricamente, este patrón fue observado por Vladimir Markovnikov en 1870, mucho antes de que se entendiera el mecanismo. La generalización que el hidrógeno agrega al carbono con más hidrógenos (otra forma de decir lo que está en negrita arriba) se conoce como regla de Markovnikov. Las aplicaciones comunes incluyen las adiciones de HBr y agua (discutidas anteriormente) y HCl.
Hidroboración-Oxidación
Observe que no podemos hacer un alcohol primario agregando agua a ningún otro alqueno que no sea etileno mediante una reacción de adición electrófila como se describió anteriormente. Una secuencia de reacción alternativa que sí logra este resultado fue desarrollada hace unos 40 años por H. C. Brown. Se esboza de la siguiente manera.
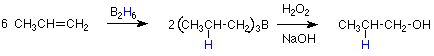
Centraremos nuestra atención en el primer paso. Conocemos el producto de este paso, por lo que podemos identificar los átomos que están actuando como electrófilo y nucleófilo. El boro se suma al carbono menos sustituido, por lo que está actuando como electrófilo. El hidrógeno se suma al carbono más sustituido, por lo que está actuando como el nucleófilo. ¿Tiene sentido esto? Hemos visto el enlace B-H actuar de esta manera antes en la reducción de cetonas y aldehídos por BH 4 - (en borohidruro de sodio). Ahí el hidrógeno actuó como nucleófilo y lo explicamos al decir que el hidrógeno es más electronegativo que el boro. Podemos entender la reacción del boro como electrófilo si consideramos que B 2 H 6 se disocia en dos moléculas de BH 3. Una comprobación rápida de la configuración electrónica de BH 3 muestra que el boro tiene un orbital 2p vacío, por lo que BH 3 tiene las características estructurales necesarias para calificarlo como electrófilo.
Prácticamente, el resultado general de esta secuencia de reacción es agregar agua a un doble enlace alqueno en una orientación que es opuesta a la predicha por la regla de Markovnikov. Por esa razón se le conoce como una adición “anti-Markovnikov”. Es un método potente para hacer alcoholes primarios y otros alcoholes donde se desea ubicar el grupo OH en el átomo de carbono menos sustituido.
Adición de halógenos
Las moléculas halógenas como Cl 2 y Br 2 también agregan al alqueno dobles enlaces. Aquí no necesitamos preocuparnos por la orientación ya que los dos extremos de la molécula adidora son idénticos, pero el mecanismo de adición electrófila nos ayuda a comprender otra característica de esta reacción, su estereoquímica.
Si se agrega bromo al ciclopenteno, podríamos anticipar dos productos que difieren en cómo los átomos de bromo están geométricamente relacionados entre sí. Si los dos bromo están en la misma cara del anillo, el compuesto se llama cis. Si están en caras opuestas, el compuesto se llama trans. El resultado experimental es que solo se forma el producto trans.
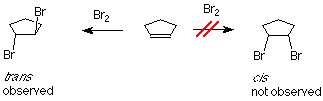
Uno de los átomos de bromo está actuando como electrófilo. Si aplicamos el mecanismo habitual, es el primer paso iría así:
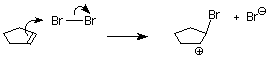
Como aprendimos en nuestro estudio de las reacciones de S N 1, los carbocationes son atacados por nucleófilos en ambas caras. Si un carbocatión está presente en este sistema, esperaríamos encontrar tanto los productos cis como los trans.
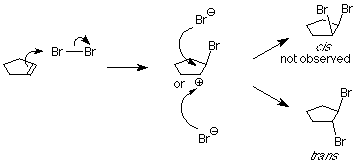
Esto no es lo que sucede cuando se realiza el experimento, por lo que concluimos que el carbocatión no está presente. Algo más está sucediendo, algo que impide que el ion bromuro nucleofílico ataque la cara del carbocatión que ya está unido al primer bromo. Entendemos eso imaginando que el átomo de bromo electrófilo se adhiere a ambos átomos de carbono alquenos. Un enlace se hace usando los electrones del enlace pi, y el otro se hace usando un par de electrones no compartidos del bromo. Esto da como resultado un nuevo anillo formado a partir del bromo y los dos átomos de carbono de alqueno. Se le llama ion “bromonio”.
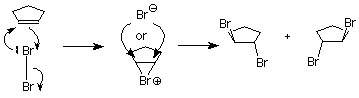
El ataque por el ion bromuro nucleofílico sobre el bromo en este anillo solo daría como resultado ciclopenteno y bromo, por lo que no se produciría ninguna reacción. El ataque por el ion bromuro sobre cualquiera de los carbonos del alqueno sería como una reacción de S N 2. El carbono atacado se invertiría, y el producto tendría la configuración trans. (Observe que existen dos productos de este tipo, que son enantiómeros, por lo que obtenemos una mezcla racémica.)
Hidrogenación
Hay otra reacción de alquenos, la hidrogenación, que merece mención pero que no está relacionada con el mecanismo de adición electrófila. La hidrogenación es la adición de hidrógeno molecular (H 2 2) al doble enlace alqueno. Esto convierte un alqueno simple en un alcano.
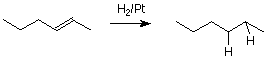
Las reacciones de hidrogenación se llevan a cabo en presencia de un catalizador sólido tal como platino (Pt) metal finamente dividido. La reacción ocurre en la superficie del metal e implica la unión temporal tanto del alqueno como de la molécula de hidrógeno a los átomos en la superficie metálica.
La hidrogenación es importante en la conversión de grasas altamente insaturadas (muchos dobles enlaces) en grasas con menos enlaces dobles. Esto se hace para que el producto tenga un punto de fusión más alto que el reactivo y sea más conveniente como untable dietético para pan. Además, los productos son menos susceptibles a la reacción con el oxígeno y no se vuelven rancios tan rápidamente.