
1.31: Sustitución electrofílica
- Page ID
- 74232

Electrofilos y Productos
Comencemos recordando los pasos clave en un mecanismo de sustitución aromática electrofílica.
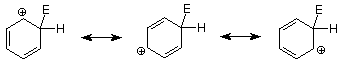
Una característica importante de este mecanismo es que podemos identificar el electrófilo si conocemos el producto porque es el átomo o grupo el que sustituye al H +. Por el contrario, si conocemos el electrófilo, podemos predecir la estructura del producto. La siguiente tabla describe estas relaciones con más detalle para varias reacciones que siguen la ruta de sustitución aromática electrofílica.
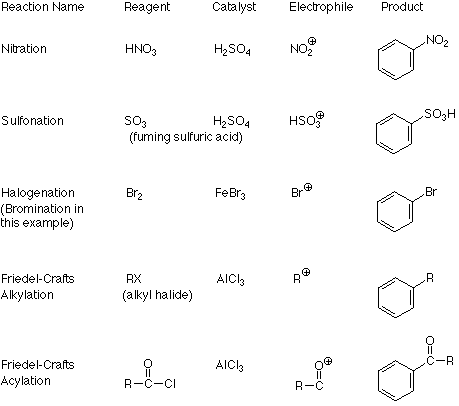
La conexión entre el electrófilo y el producto queda clara a partir de estos ejemplos. Lo que no está claro es cómo el catalizador transforma el reactivo en el electrófilo. Aquí hay dos patrones. Una se aplica cuando el electrófilo se hace eliminando un ion haluro del reactivo (halogenación y las dos reacciones de Friedel-Crafts). La formación de un carbocatión a partir de un haluro de alquilo y tricloruro de aluminio es un ejemplo típico del primer proceso:
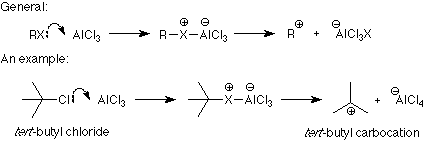
Observe que el papel del catalizador es vincularse con el grupo de salida y convertirlo en un mejor grupo de salida. Esto hace que la formación del electrófilo (carbocatión en este caso) sea mucho más fácil para que haya más electrófilo alrededor para atacar el anillo de benceno. Mecanismos similares se utilizan en las reacciones de halogenación y acilación de Friedel-Crafts.
Para la sulfonación y nitración, el catalizador es ácido sulfúrico. Así como fue el caso en las reacciones S N 1 y S N 2 de los alcoholes, la función del ácido es protonar un par de electrones no compartidos sobre el oxígeno. Cuando está involucrado el grupo OH del ácido nítrico, el grupo OH 2 + que se forma es un buen grupo saliente. Su salida hace que el electrófilo activo:
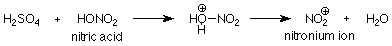
En resumen, la función del catalizador es convertir el reactivo en un electrófilo fuerte, que luego ataca los electrones pi en el anillo aromático para formar un nuevo enlace covalente. La reacción se completa con la pérdida de un H +.
Efectos Directivos Ortho-Para
El patrón reactivo - catalizador - electrófilo - producto funciona bien cuando el compuesto aromático es benceno en sí, pero las cosas se complican más cuando se utiliza un benceno sustituido (una molécula en la que uno de los hidrógenos del benceno ha sido reemplazado por otro átomo o grupo). Hay tres productos que pueden surgir en dicho sistema. Como ejemplo echemos un vistazo al tolueno —que es metilbenceno. Usaremos la nitración como nuestro ejemplo de sustitución aromática electrofílica aquí.
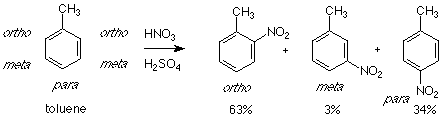
Aunque hay cinco hidrógenos disponibles para ser reemplazados en el anillo bencénico del tolueno, dos de ellos son directamente adyacentes (orto) al grupo metilo. El ataque en cualquiera de estas posiciones da el mismo producto. Consideraciones similares se aplican a la meta posición. El resultado es que sólo se obtienen tres productos. La distribución real de estos productos muestra que se obtiene muy poco del meta producto. En cambio, casi todo el producto consiste en los isómeros orto y para. Por esta razón el grupo metilo se denomina “grupo orto-para director”. “Dirige” al electrófilo entrante para que ataque en las posiciones orto y para relativas a sí mismo.
¿Cómo ejerce el grupo metilo este efecto “directivo”? Podemos comenzar a darle sentido al recordar que explicamos la regla de Markovnikov considerando que el producto que es más abundante se forma más rápido a través de una vía que tiene la menor energía de activación. Eso nos lleva a una consideración de las energías relativas de los estados de transición competidores y al uso de intermedios como “suplentes” útiles para esos estados de transición. Concluimos que la regla de Markovnikov se explica por la idea de que los productos más abundantes están formados por vías que pasan por intermedios de menor energía.
Si aplicamos el mismo razonamiento a estos efectos directivos, la pregunta se convierte en “¿Qué hace que los intermedios para la formación de orto y para productos sean más estables que los de los productos para? Como es habitual, buscaremos las respuestas comparando las estructuras de los intermedios.
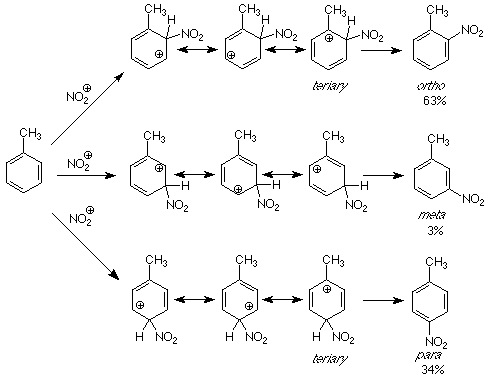
Cada uno de los intermedios se describe mediante un híbrido de resonancia que incluye tres carbocationes. Sin embargo, para los intermedios que conducen a los productos orto y para, una de las estructuras carbocatiónicas contribuyentes es terciaria. Esta estructura es más estable que las otras porque los electrones en el grupo metilo pueden estabilizar directamente el carbono carbocatiónico deficiente en electrones. Esta estabilidad se transmite al híbrido de resonancia, lo que hace que los intermedios para el ataque sean más estables que los del ataque en la posición meta. Los intermedios más estables significan estados de transición de energía más bajos y reacciones más rápidas. Una reacción más rápida significa que se forma más producto a través de esa vía.
El resultado de este análisis es que los grupos que pueden donar electrones estabilizan los carbocationes. El ataque electrófilo será más rápido en posiciones tales que los carbocationes producidos tienen cargas positivas en los carbonos que están unidos a grupos donadores de electrones como los grupos methy. Esto ocurre cuando el ataque ocurre en las posiciones orto y para al grupo donador de electrones. Dicha donación de electrones ocurre con grupos alquilo en general y cuando átomos como el oxígeno y el amino-nitrógeno son atacados directamente al anillo. En estos últimos casos es el par no compartido sobre el oxígeno o nitrógeno el que ayuda a reducir la energía del átomo de carbono deficiente en electrones. (Vea la figura en la parte inferior de p 165 en Atkins & Carey para un ejemplo.) Examinar los grupos orto-para directores en la Tabla 6.3 (p 162 de Atkins & Carey) y decidir cómo actúa cada uno como un grupo donador de electrones.
Efectos de la Meta Directiva
¿Podemos extender este análisis a grupos que están meta dirigiendo? Vamos a probar un ejemplo. En la nitración de benzaldehído la mezcla de productos es 19% orto, 72% meta, y 9% para. El grupo aldehído carbonilo (y los grupos carbonilo en general) es un sustituyente meta director. Veremos las estructuras de resonancia del intermedio para una explicación:
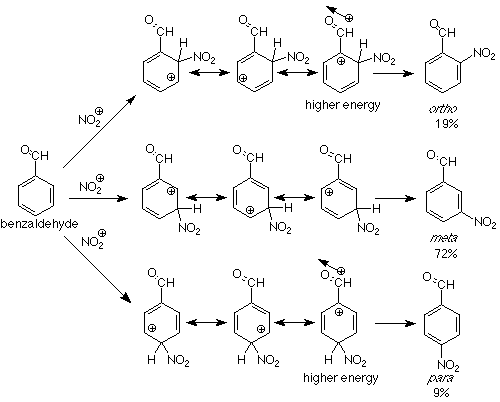
Vemos que el ataque en las posiciones orto y para al grupo carbonilo coloca la carga positiva directamente adyacente al extremo positivo del dipolo del grupo carbonilo. La energía de dicho intermedio será mayor que aquella en la que la carga positiva nunca se aproxime al carbono carbonílico, lo que es el caso en el intermedio para el metaataque. La consecuencia es que el ataque en la posición meta es más rápido y se forma más meta producto. Los grupos en los que el átomo unido directamente al anillo de benceno tienen una carga positiva parcial o completa tienden a atraer electrones hacia sí mismos. Dichos grupos se denominan grupos extractores de electrones y son grupos meta directores en sustitución aromática electrofílica. Fíjese en los grupos meta directores en la Tabla 6.3 (p 162 en Atkins & Carey) y vea si se confirma la declaración anterior.
Activación y Desactivación
La idea subyacente en el análisis anterior de los efectos directivos en electrofílicos fue comprender las velocidades de reacción relativas cuando la competencia era entre diferentes sitios en una misma molécula. Esto se denomina competencia intramolecular y también se utilizó en nuestra comprensión de la regla de Markovnikov. También podemos observar la competencia entre moléculas —competencia intermolecular— usando las mismas ideas, comparando las energías de los intermedios en vías competidoras. Por ejemplo, imaginemos una reacción en la que mezclamos tolueno y benceno y luego hacemos una nitración. Si obtenemos más nitrotolueno (los tres isómeros) que nitrobenceno, el tolueno ha reaccionado más rápido que el benceno. Decimos que el grupo metilo “activó” el anillo de benceno. Entendemos que esto significa que el grupo metilo disminuyó la energía de los intermedios y estados de transición a lo largo de la ruta al producto en comparación con el efecto de un hidrógeno (en benceno).
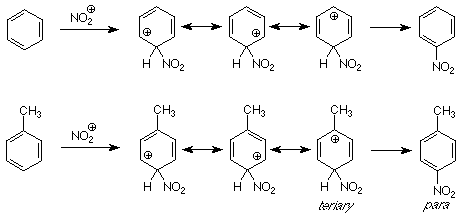
Una comparación de los intermedios a lo largo de las rutas del benceno y el tolueno a sus respectivos productos nos muestra la razón. (Solo se muestra la vía para; la vía orto es similar). El intermedio para ataque para al grupo metilo incluye una estructura de resonancia que es terciaria y por lo tanto es estabilizada por el carácter donador de electrones del grupo metilo. No existe tal estructura en el intermedio para la nitración del benceno, por lo que el intermedio para la nitración del tolueno es más estable y la reacción que lo atraviesa es más rápida. Llamamos al grupo methy (y a los grupos alquilo en general) un grupo “activador” para la sustitución aromática electrófila.
Dado que las características donadoras de electrones del grupo metilo son responsables tanto de sus propiedades activantes como de sus propiedades de dirección orto-para, podríamos sospechar que los grupos orto-para directores también serán grupos activadores. Un examen de la Tabla 6.3 (p 162, Atkins & Carey) muestra que esto es generalmente cierto. (Las excepciones, los halógenos, se tratan en la Sección 6.10 de Atkins & Carey. No los discutiremos más)
Esta conclusión también sugiere que los grupos meta directores también serán grupos “desactivantes”; es decir, que los anillos de benceno que llevan un grupo meta director serán menos reactivos que los que no lo hacen. El Cuadro 6.3 también lo demuestra. Tanto las características meta directoras como las características de desactivación son el resultado de la naturaleza de extracción de electrones de estos grupos y el consiguiente aumento en la energía de los intermedios de sustitución aromática electrofílica cuando están presentes.