
1.32: Oxidaciones de cadenas laterales, fenoles, arilaminas




- Última actualización
- 30 oct 2022
- Guardar como PDF

( \newcommand{\kernel}{\mathrm{null}\,}\)
Carbocationes estabilizados por resonancia
Comencemos por observar la adición de HCl a un alqueno en el que uno de los carbonos del alqueno está unido directamente al anillo de benceno.
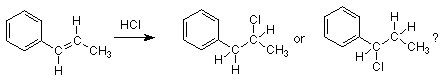
En este ejemplo cada uno de los carbonos alquenos tiene el mismo número de hidrógenos por lo que se bloquea la aplicación directa de la regla de Markovnikov. Encontramos que debemos ir más allá de la regla de Markovnikov hasta su explicación en términos de la relativa estabilidad de los dos carbocationes que podrían formarse. Aquí están los dos carbocationes que se formarían por el ataque de H + sobre el alqueno:
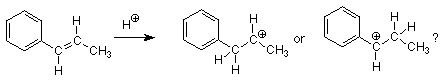
¿Cuál de estos carbocationes tiene la menor energía? ¿El grupo fenilo donará electrones de manera más efectiva que el grupo metilo? Lo clave a notar al responder a estas preguntas es que los electrones pi del anillo de benceno están directamente adyacentes al carbono carbocatión en la estructura de la izquierda. Esto es una reminiscencia de la situación que vimos en la adición de HCl al 1,3-butadieno donde encontramos estabilización de resonancia del carbocatión por los electrones pi del doble enlace vecino. La misma situación ocurre aquí, y podemos simbolizar la estabilización de resonancia con las siguientes estructuras de resonancia.
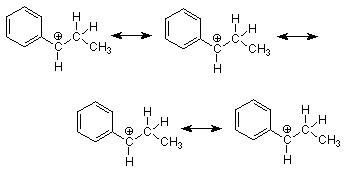
El efecto de esta resonancia es hacer que el carbocatión sea más estable cuando la carga y la deficiencia de electrones se localizan sobre un carbono que está unido directamente al grupo fenilo. Tal carbono se llama carbono “bencílico”. Dado que no existe tal estabilización de resonancia disponible cuando un grupo alquilo simple se une al carbono carbocatiónico, el carbocatión bencílico es más estable, se forma más rápido, y el producto que se forma a partir de él es el único que vemos.
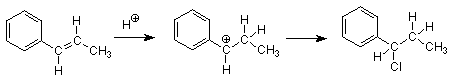
Con estos antecedentes, podemos decidir rápidamente cuál de los dos haluros de alquilo inferiores reaccionará más rápido por medio de un mecanismo S N 1.
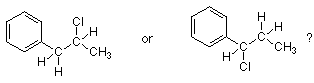
El haluro de alquilo más reactivo es el que da el carbocatión más estable. El carbocatión más estable (ya que ambos serán carbocationes secundarios) es aquel donde es posible la resonancia bencílica. (Las estructuras de resonancia se dan anteriormente.) Solo el haluro de alquilo de la derecha producirá un carbocatión en el que el carbono del carbocatión está unido directamente a un grupo fenilo. En consecuencia entendemos por qué el haluro de alquilo de la derecha es más reactivo.
Bases estabilizadas por resonancia
Hemos visto que los carbocationes bencílicos se estabilizan por resonancia. ¿Qué sucede si el átomo directamente unido al anillo de benceno tiene un par de electrones no compartidos? En otras palabras, es rico en electrones en lugar de pobre en electrones. Podemos ver los efectos de este arreglo en las propiedades ácido-base de fenoles y aminas aromáticas.
Un fenol es un alcohol cuyo grupo R es un grupo fenilo. Es esencial que el oxígeno y el anillo de benceno estén unidos directamente entre sí. Si hay un carbono tetraédrico entre ellos, el compuesto no es un fenol sino un alcohol.

Una diferencia importante entre fenoles y alcoholes radica en sus acidedades. Recordemos que los alcoholes, como el agua, tienen p K a's de aproximadamente 16. Por el contrario, los fenoles suelen tener p K a de aproximadamente 10, lo que los convierte en ácidos considerablemente más fuertes que los alcoholes. Dado que el ácido más fuerte tiene la base conjugada más débil, necesitamos preguntarnos por qué un fenóxido (la base conjugada de un fenol) es más débil que un alcóxido (la base conjugada de un alcohol).
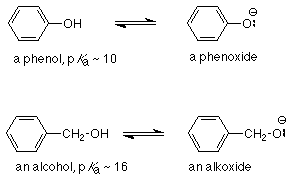
Dado que es el par de electrones no compartidos el que hace que cualquiera de las bases sea una base, podemos preguntarnos por qué el par de electrones no compartidos en el ion fenóxido está menos disponible para enlazarse con un protón que el par de electrones no compartidos en el ion alcóxido. Nuevamente, la razón se encuentra en la resonancia.
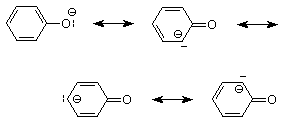
Las estructuras de resonancia que mejoran la descripción del ion fenóxido muestran que el par de electrones en el oxígeno se mueve parcialmente a una posición de enlace pi entre el oxígeno y el carbono de fenilo al que está unido. En esa medida, el par de electrones en un ion fenóxido está menos disponible para la unión a un protón y el ion fenóxido es una base más débil que un ion alcóxido en el que no es posible tal resonancia.
La misma situación ocurre cuando un nitrógeno amino está conectado directamente a un anillo aromático.
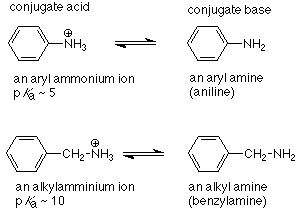
Vemos que el ion amonio en el que el nitrógeno está unido directamente al grupo fenilo es un ácido más fuerte que aquel en el que el nitrógeno está unido a un átomo de carbono hibridado sp 3. Esto significa que la base conjugada, la aril amina, en la que el nitrógeno está unido al grupo fenilo también es más débil que su homólogo de alquilamina. Esto se entiende por resonancia entre el par de electrones no compartidos sobre nitrógeno y el anillo aromático, solo posible si la conexión es directa, lo que hace que el par de electrones no compartidos esté menos disponible para servir como base.
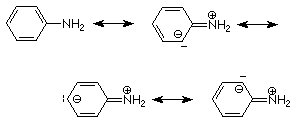
Las posibilidades de resonancia para un grupo aromático le permiten servir como sustituyente donador de electrones cuando el átomo unido necesita electrones (carbocatión) o como sustituyente aceptor de electrones cuando el átomo unido tiene un par no compartido para compartir (oxígeno o nitrógeno).
Oxidación de cadena lateral
Hay otra reacción que ocurre en el átomo directamente unido a un anillo aromático. Esto se conoce como “oxidación de cadena lateral”. Cuando un compuesto que tiene un grupo alquilo directamente unido a un grupo arilo se trata con un agente oxidante fuerte como el ácido crómico, el carbono bencílico se oxida a un grupo ácido carboxílico que permanece unido al grupo arilo. Cualquier otro enlace carbono-carbono se rompe. Hay una restricción más: el carbono bencílico debe tener un hidrógeno unido.
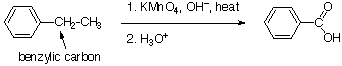
El mecanismo de esta reacción es oscuro, pero el hecho de que específicamente requiera que haya un enlace C-H bencílico sugiere que romper este enlace es esencial. Cualquier intermedio que pueda formarse rompiendo este enlace se estabilizará por resonancia con el grupo arilo, lo que proporciona una explicación de la especificidad del ataque en la posición bencílica. Tales reacciones también ocurren en un contexto biológico. Las enzimas oxidan las cadenas laterales de alquilo en anillos aromáticos como parte de hacer que dichos compuestos sean lo suficientemente solubles como para ser eliminados.
Iones Diazonio
Cuando las aril aminas primarias reaccionan con ácido nitroso (HNO 2, generado a partir del nitrito de sodio (NaNO 2 y HCl) se produce una reacción que hace un nuevo triple enlace nitrógeno-nitrógeno. No nos preocuparemos por el mecanismo, pero el producto (llamado ion diazonio) es un valioso intermedio sintético.
El ion diazonio rara vez se aísla (los iones diazonio pueden ser explosivos) por lo que en su lugar se usa en la solución en la que se elabora. El grupo N 2 puede ser reemplazado por una variedad de otros grupos y también puede ser un electrófilo en sí mismo hacia un anillo aromático suficientemente activado. Las posibilidades se describen en la siguiente tabla:
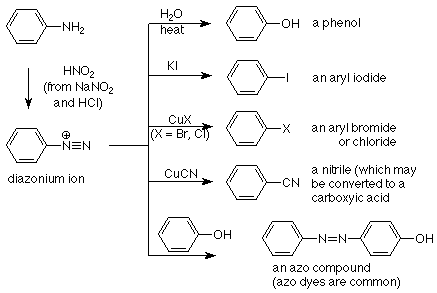
Las primeras cuatro reacciones de esta tabla son ejemplos de la sustitución del grupo N 2 por un nucleófilo. Estas reacciones proporcionan una manera de realizar sustituciones nucleofílicas en un carbono de un anillo aromático que no son posibles por mecanismos normales de S N 1 y S N 2. La gran estabilidad del nitrógeno molecular (N 2) lo convierte en un grupo de salida muy bueno, pero los mecanismos de estas reacciones no son simples.
La última reacción en la tabla es aquella en la que el grupo diazonio actúa como electrófilo. No es un electrófilo particularmente bueno, por lo que requiere un anillo reactivo como objetivo. Tal anillo necesita tener un fuerte grupo donador de electrones como OH o NH 2 para aumentar su reactividad en un mecanismo de sustitución aromática electrofílica.
El producto del acoplamiento electrófilo de un ion diazonio con un anillo aromático reactivo incluye un doble enlace nitrógeno-nitrógeno. Estos compuestos suelen ser intensamente coloreados y son útiles como colorantes. Se puede incluir una variedad de otros grupos funcionales, que a menudo llevan cargas iónicas, para que se adhieran más a la tela. A menudo se utilizan términos como tintes de anilina o tintes azoicos para referirse a estos compuestos.
Las sales de diazonio son materiales intermedios sintéticos versátiles y sabemos que están hechas de anilina o moléculas similares. ¿De dónde provienen la anilina y sus análogos? La vía más común es por reducción de nitrobenceno o nitrobencenos sustituidos. El agente reductor habitual es el estaño (en el laboratorio) o el hierro (en la práctica industrial) utilizado en solución acuosa de HCl. Dado que la nitración es una ruta de uso común al nitrobenceno, la secuencia habitual para la producción de anilina es:
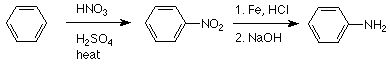
El uso de NaOH en la etapa final es para neutralizar el HCl de manera que la amina básica (que de otro modo estaría en forma de su ion amonio ácido conjugado) sea lo suficientemente no polar para ser extraída del agua.