
4.7: Biosíntesis y Síntesis Total de Esteroides
- Page ID
- 70272

Biosíntesis de Linosterol
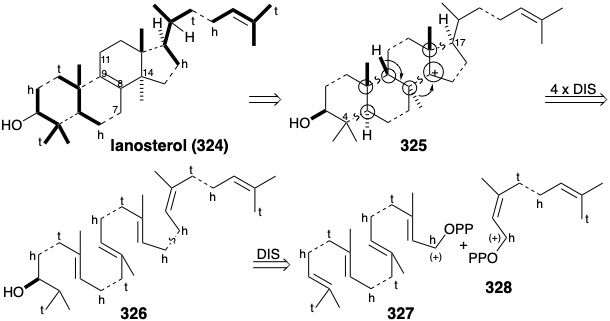
Lanosterol (324) es un miembro de la familia de esteroides de productos naturales. El origen isoprenoide de este triterpeno, biogenéticamente un hexámero C 30 de pirofosfato de isopentenilo, no es del todo evidente a partir de su estructura. Así, mientras que dos unidades de isopreno son discernibles en la porción derecha de 324 y dos en la porción izquierda, los diez átomos de carbono en la región central de la molécula no muestran conectividad isoprenoide. Sin embargo, si se anexara un grupo metilo en la posición 8 o 9 de la cadena de cuatro carbonos 7,8,9,11 se formaría una unidad de isopreno. Dado que la generación de las redes de carbono multicíclicos de terpenos se produce por ciclaciones de polieno inducidas por electrófilos, el enlace 8,9-C=C en 324 podría surgir por la eliminación de un protón de un precursor de carbocatión. Un carbocatión en la posición 8 podría haber sido generado por la migración 1,2-de un grupo metilo desde la posición 8 a un carbocatión en la posición 14. Dos unidades más de isopreno son discernibles en la porción central del precursor putativo 325. El análisis topológico de 325 revela seis átomos comunes. Dos desconexiones entre átomos comunes y dos entre un átomo común y otro no común simplifican enormemente la estructura sugiriendo un precursor triterpénico acíclico 326 que podría llegar por la unión cabeza a cabeza de un pirofosfato diterpénico 327 con un pirofosfato monoterpénico 328. La estrategia inferida anteriormente es cercana a la involucrada en la biosíntesis del lanosterol.
La estrategia biosintética real implica de hecho una unión cabeza a cabeza de pirofosfatos terpenoides. Sin embargo, se logra una construcción más eficiente a través de un intermedio triterpénico acíclico simétrico, escualeno (333), formado por la unión de dos moléculas de un precursor de sesquiterpeno, E, pirofosfato de E-farnesilo (8). Dado que ambos carbonos a unir son electrófilos, la formación polar del enlace C-C central del escualeno no puede ser directa. 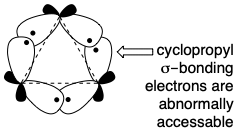 Más bien, solo se utiliza la activación electrófila proporcionada por el grupo funcional de una molécula de farnesil-OPP para formar un enlace C-C con el enlace nucleofílico C=C de una segunda molécula de farnesil-OPP. La pérdida de protones del supuesto intermedio 3° carbocatión 329 (o, posiblemente, el aducto correspondiente con un resto nucleofílico de la enzima que cataliza el proceso) produce un intermedio ciclopropilcarbinilo, pirofosfato de presqualeno (330), que puede aislarse de biosíntesis realizadas in vitro en ausencia de NADPH. En presencia de NADPH, 330 se somete a un reordenamiento reductivo que involucra formalmente un intermedio catiónico ciclopropil 3° carbinilo reordenado 331 y un catión alílico 332 que es capturado por hidruro para suministrar escualeno (333). El enlace σ-en el pirofosfato de ciclopropil-carbinilo 330 sirve como nucleófilo que desplaza un nucleófugo de pirofosfato. Los electrones de enlace σen los ciclopropanos son especialmente accesibles.
Más bien, solo se utiliza la activación electrófila proporcionada por el grupo funcional de una molécula de farnesil-OPP para formar un enlace C-C con el enlace nucleofílico C=C de una segunda molécula de farnesil-OPP. La pérdida de protones del supuesto intermedio 3° carbocatión 329 (o, posiblemente, el aducto correspondiente con un resto nucleofílico de la enzima que cataliza el proceso) produce un intermedio ciclopropilcarbinilo, pirofosfato de presqualeno (330), que puede aislarse de biosíntesis realizadas in vitro en ausencia de NADPH. En presencia de NADPH, 330 se somete a un reordenamiento reductivo que involucra formalmente un intermedio catiónico ciclopropil 3° carbinilo reordenado 331 y un catión alílico 332 que es capturado por hidruro para suministrar escualeno (333). El enlace σ-en el pirofosfato de ciclopropil-carbinilo 330 sirve como nucleófilo que desplaza un nucleófugo de pirofosfato. Los electrones de enlace σen los ciclopropanos son especialmente accesibles.

La epoxidación asimétrica convierte el precursor proquiral del triterpeno acíclico 333 en un epóxido homoquiral, óxido de escualeno (334). La generación de los cuatro anillos fusionados de lanosterol se inicia entonces por alquilación intramolecular de un enlace C=C por un electrófilo terciario proporcionado por la protonación del epóxido. Un total de cuatro alquilaciones consecutivas, conocidas como ciclación de polieno, entregan el supuesto intermedio 335 que se reorganiza por una serie de dos desplazamientos de 1,2-hidruro y dos 1,2-metilo para dar lanosterol (324) después de la pérdida de protones desde la posición 9.
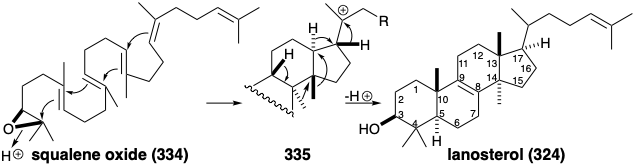
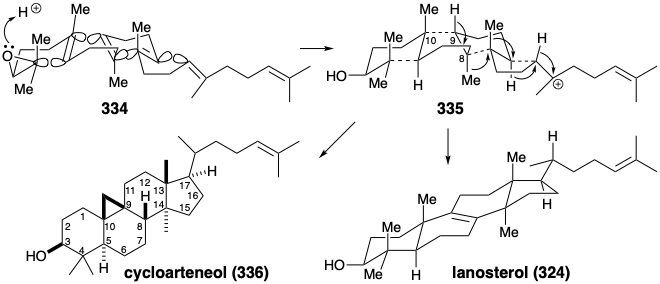
Especialmente interesantes son los detalles estereoquímicos de la ciclación del polieno y posterior reordenamiento de 335 para entregar 324. La ciclación del polieno implica la adición antiperiplanar estereoespecífica a través de tres enlaces C=C en 334 (ver más abajo). La conformación plegada de 334, requerida para la ciclación a 335, es probablemente impuesta por la enzima involucrada ya que la congestión estérica apreciable está presente tanto en 334 como en el intermedio 335. El alivio de la tensión estérica proporciona una gran fuerza impulsora para el reordenamiento de 335 a 324 que implica la inversión estereoespecífica de la configuración en cada estereocentro durante los desplazamientos de 1,2-hidrógeno o metilo.  Así, cada desplazamiento de 1,2-hidruro o 1,2-metilo ocurre en la parte posterior del orbital conectado al nucleófugo saliente en lo que puede verse como una serie de reacciones de sustitución nucleofílica, donde los pares de electrones de enlace σsirven como nucleófilos. En lugar de la pérdida de protones para dar 324, el reordenamiento de 334 en algunas plantas y algas superiores termina con la migración de protones de la posición 9 a la 8 y la pérdida de protones del grupo metilo en la posición 10 formando un anillo de ciclopropano en el cicloarteneol (336). Este mecanismo para la generación de un ciclopropano es análogo al de la producción de pirofosfato de presqualeno (330) a partir del 329.
Así, cada desplazamiento de 1,2-hidruro o 1,2-metilo ocurre en la parte posterior del orbital conectado al nucleófugo saliente en lo que puede verse como una serie de reacciones de sustitución nucleofílica, donde los pares de electrones de enlace σsirven como nucleófilos. En lugar de la pérdida de protones para dar 324, el reordenamiento de 334 en algunas plantas y algas superiores termina con la migración de protones de la posición 9 a la 8 y la pérdida de protones del grupo metilo en la posición 10 formando un anillo de ciclopropano en el cicloarteneol (336). Este mecanismo para la generación de un ciclopropano es análogo al de la producción de pirofosfato de presqualeno (330) a partir del 329.
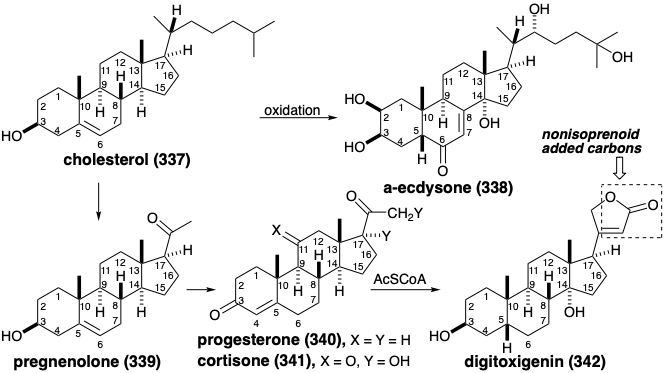
Nor y Seco Esteroides. Aunque el lanosterol (324) y el cicloarteneol (336) son triterpenos irregulares, es decir, sus esqueletos de carbono no están compuestos por unidades de isopreno intactas, estos triterpenos poseen los treinta carbonos esperados. Muchos esteroides, que se derivan biológicamente del lanosterol, contienen menos de treinta carbonos y son referidos como ni triterpenos. Así, por ejemplo, la formación de colesterol (337) a partir del lanosterol (324) ocurre por conversión oxidativa de tres grupos metilo en sustituyentes formilo o carboxilo que se pierden como formiato o dióxido de cabón para dar un tris ni triterpeno después de la saturación del enlace C=C de la cadena lateral y migración del enlace Δ 8,9 C=C a la posición Δ 5,6. La biosíntesis de algunos esteroides a partir del colesterol, como la hormona de muda de insectos α-ecdisona (338), implica simplemente la introducción oxidativa de funcionalidad y migraciones de enlaces C=C. La pérdida de seis carbonos de la cadena lateral del colesterol (337) lleva a pregnenolona (339) el precursor biosintético de la hormona reproductiva femenina progesterona (340) y las hormonas adrenocorticales como la cortisona (341) que es generada por funcionalización oxidativa de 340. La biosíntesis de los esteroides cardíacos como la digitoxigenina (342), que ocurre en las plantas, crea el resto butenólido mediante la adición de un nucleófilo de dos carbonos de acetilCoA al carbonilo electrófilo de cadena lateral de 340.
La adición de un átomo de carbono como grupo metilo electrófilo a partir de S-adenosilmetionina (SAM) ocurre durante la biosíntesis del ergosterol (343). Un reordenamiento pericíclico de 343 a 344 seguido de un reordenamiento antarafacial [1.7] -sigmatropico térmicamente permitido del hidrógeno desde el metilo en la posición 10 a la posición 9 conduce a la generación de vitamina D 2 (345). Los desplazamientos sigmatrópicos del hidrógeno necesariamente implican solapamiento positivo, es decir, con la misma fase, del hidrógeno semiocupado σ-orbital con los extremos del pentadienilo n-orbital ocupado más alto Tanto 344 como 345 son esteroides secos, es decir, sus esqueletos de carbono carecen de uno de los enlaces C-C del anillo del esqueleto esteroideo tetracíclico.

Toda la cadena lateral del colesterol se elimina durante la biosíntesis de las hormonas sexuales masculinas androsterona (346) y testosterona (347) y la biosíntesis de la hormona reproductiva femenina estrona (348) incluso requiere la pérdida del sustituyente metilo angular desde la posición 10. Cabe destacar que la estrategia biosintética para 348 es excepcionalmente sinuosa y larga considerando la simplicidad estructural (solo cuatro centros de quiralidad) de esta diana. La justificación, por supuesto, es la disponibilidad del material de partida, el omnipresente precursor biológico esteroide, el colesterol. Además, la naturaleza tiene a su disposición un vasto armementario de reactivos selectivos (enzimas) para lograr la eliminación quirúrgicamente limpia de átomos o grupos de carbono no deseados mediante la activación de enlaces C-H, incluso aquellos que están alejados de la funcionalidad.
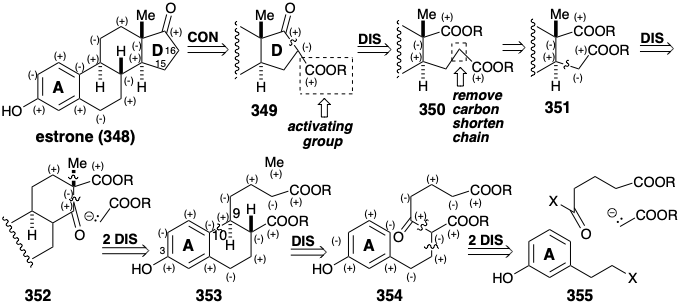
Síntesis Total de Estrona. Todos los circuitos entre la funcionalidad oxígeno en los anillos A y D de la estrona son consonantes excepto aquellos que involucran los carbonos 15 y 16. Por lo tanto, la funcionalidad activadora es esencial para permitir la formación de enlaces C-C polares entre el carbono carbonilo del anillo D y el carbono 16. La desconexión de este enlace en 349 sugiere un diéster 350 que todavía tiene disonancia funcional, por ejemplo entre los dos grupos carboxilo. Toda disonancia se elimina acortando el propiónico a una cadena lateral acética como en 351. La desconexión polar de un nucleófilo estabilizado con éster sugiere un β-ceto-éster 352 y otras desconexiones polares sugieren 353 y un metil electrófilo, como precursores. Finalmente, la desconexión polar de 353 sugiere un precursor aromático monocíclico 354 con una cadena lateral completamente consonante en la que el carbonilo agregado en la posición 9 incipiente puede facilitar desconexiones polares adicionales a 355, un nucleófilo estabilizado con éster, y un glutaryl electrófilo. El análisis retrosintético anterior proporciona una estrategia lineal para la síntesis de estrona que comienza con un anillo A intacto y luego construye los anillos B, C y D sucesivamente por reacciones polares.
La primera síntesis total de estrona, se completó 21 en 1948. Se ensambló un metil éter 357, relacionado con 354, a partir del bromoetilanisol (356) por alquilación y luego acilación de malonato de dietilo. La ciclalquilación del cetotriéster resultante 357 hace uso de la activación polar proporcionada por la funcionalidad relacionada con la diana en la posición 3 que se conjuga a través del anillo aromático con la posición 10. La hidrólisis y eliminación descarboxilativa luego liberaron el alqueno 358 22 que tras la hidrogenación, O-metilación, ciclación de Dieckman y C-metilación entregaron una mezcla epimérica a partir de la cual se aisló el cetoéster requerido 360 por cristalización fraccionada. Una vez más la funcionalidad relacionada con el objetivo, aquí el carbonilo incipiente en la posición 17, facilita la formación de enlaces polares. También es de destacar que la funcionalidad objetivo no relacionada, un grupo carbonilo en el carbono que se convertirá en la posición 9, sirve como un linchpin para conectar segmentos principales en la conversión 356 a 357 y para generar la última conexión para el anillo B en el 357 a 358 conversión. La condensación de Reformatsky seguida de deshidratación e hidrogenación proporcionó el diéster 362 y un epímero del que se separó por cristalización fraccionada. La falta de estereocontrol en esta síntesis de 362 requirió tediosos aislamientos de mezclas de isómeros y resultó en un bajo rendimiento global.
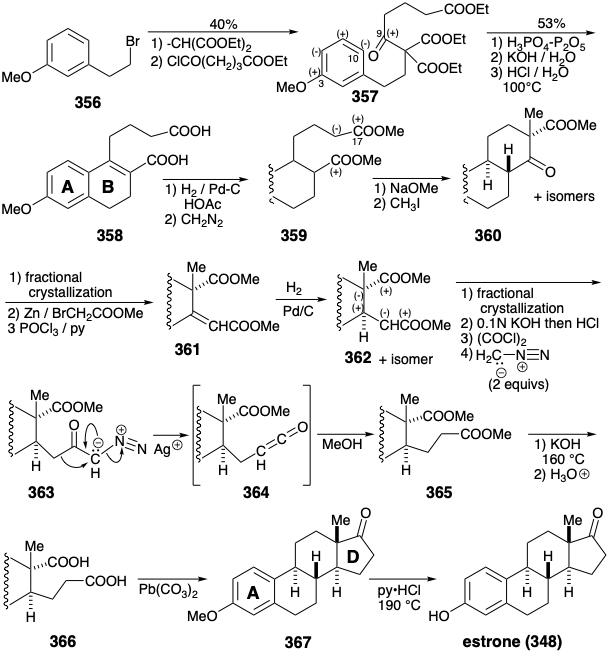
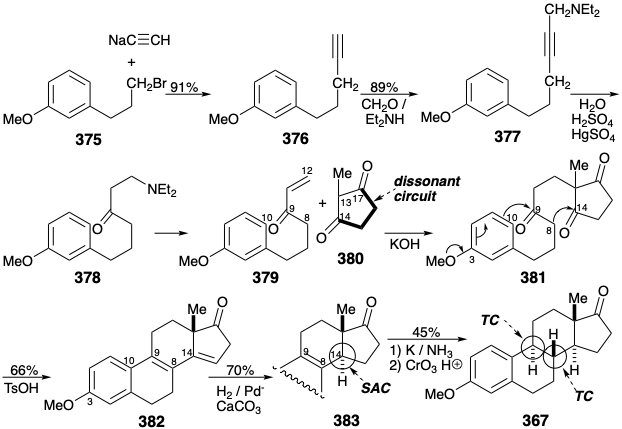
El alargamiento de cadena de la consonante 1,5-diéster 362 generó un precursor disonante de 1,6-diéster 365 del anillo D disonante ciclopentanona en 367. La creación de un producto disonante por reacciones polares requiere un reactivo disonante. En el presente caso este papel lo juega el diazometano que es disonante en virtud de la presencia de un grupo activante bifílico. Así, el grupo diazonio estabiliza un carbanión α proporcionando nucleofilia para la formación de enlaces C-C con un cloruro de acilo y posteriormente sirve como nucleófugo promoviendo la migración de un grupo alquilo nucleófilo del carbono carbonilo en 363 al carbono vecino en 364.
Una construcción alternativa del anillo D disonante ciclopentanona en 367 a partir del consonante 1,5-diéster 365 genera un enlace C-C entre los dos carbonos de éster electrófilo mediante un proceso no polar, acoplamiento reductor. 23 Así, una condensación de aciloína proporciona 368 de los cuales se elimina el carbonilo innecesario por desulfuración reductora del tioacetal derivado 369.
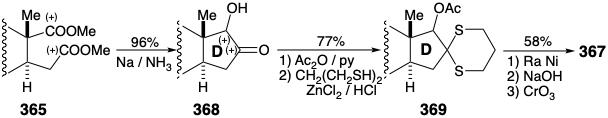
Análisis Topológico de Estrona. La estrategia topológica para una síntesis se puede resumir en un diagrama que muestra el material de partida con contorno en negrita y enlaces formados al completar el esqueleto con líneas discontinuas. Las dos síntesis de estrona anteriores ilustran estrategias que presentan la construcción tardía del anillo D disonante como en el diagrama A que permite la secuencia de conexiones C-C empleadas. Se puede lograr una mayor eficiencia mediante la incorporación del anillo D disonante como una unidad preformada como en B. Casi tan eficiente es el uso de un precursor disonante a partir del cual el anillo D se genera fácilmente inmediatamente después de ensamblar un intermedio que contiene todos los átomos de carbono requeridos para el esqueleto como en C. Una síntesis altamente convergente y, por lo tanto, excepcionalmente corta se puede lograr uniendo una unidad de anillo AB preformada con una unidad de anillo en D como en D. Las estrategias resumidas por A - D utilizan materiales de partida aromáticos para el resto aromático en la diana sintética. Esta táctica se beneficia de la estabilidad relativa de los sistemas aromáticos al evitar posibles reacciones secundarias que disminuyen el rendimiento que podrían ocurrir durante la manipulación de intermedios no aromáticos que contienen más funcionalidad reactiva. Curiosamente, algunas síntesis modernas eficientes, resumidas por E y F, generan el anillo A después del ensamblaje de un precursor no aromático que contiene todos los carbonos esqueléticos de la diana final. La discusión adicional de las estrategias B - F se aplaza a una consideración completa de cada síntesis.
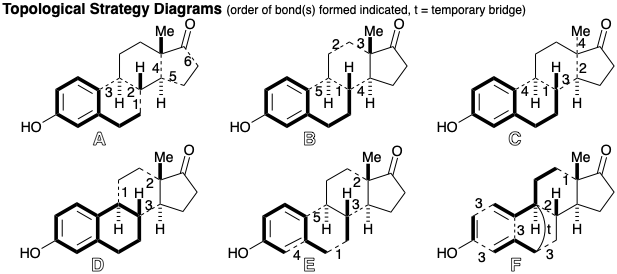
Una estrategia resumida topológicamente por B introduce la funcionalidad no relacionada con el objetivo, grupos carbonilo en 370 en los carbonos que se convertirán en las posiciones 9 y 14, para activar la unión polar de dos pares de átomos comunes y la unión polar de un nucleófilo simétrico de anillo D con 371. La dislocación de esta enona a una cetona saturada 372 con un nucleófugo (Nu) β al carbonilo y este último a un alquino 373, revela la posibilidad de explotar un nucleófilo alquino terminal para ensamblar 373 a partir de un electrófilo de arilpropilo 374.
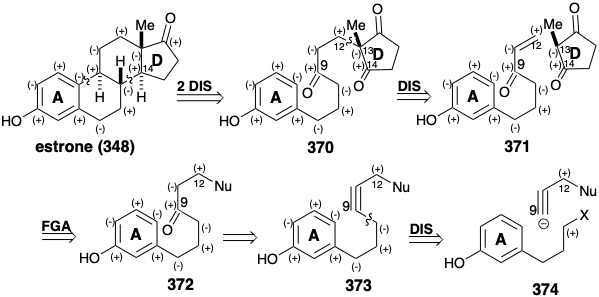
El uso de un anillo en D preformado en el bloque de construcción disonante 380 hace que la síntesis 24 sea más convergente. También se proporciona mayor eficiencia por la formación consecutiva de dos conexiones, entre las posiciones 8 y 14 y luego entre las posiciones 9 y 10, en una sola reacción que produce 382 de 381. Destaca la regioselectividad de la conversión 377 a 378. Claramente, el grupo dietilamino proporciona una influencia regiocontroladora, quizás debido a una interacción coordinativa con el\(\ce{Hg^2+}\) catalizador o a la desestabilización inductiva del desarrollo de un catión vinílico β al nitrógeno electronegativo. La generación de las relaciones estereoquímicas requeridas se logra mediante SAC durante la entrega de hidrógeno a 382 y TC durante la protonación de las conformaciones preferidas de intermedios aniónicos en la reducción de 383.
Otra síntesis más reciente, 25 resumida topológicamente por C, está estrechamente relacionada con el enfoque topológico B discutido anteriormente. Así, la topología y reactividad polar implicada en la conversión 381 a 382 es la misma que en la conversión 387 a 388. Sin embargo, no se explota un anillo en D preformado. Más bien, este resto está presente en forma latente en 386 que contiene un circuito disonante entre grupos 1,4-dicarbonilo enmascarados. La disonancia deriva de una matriz latente de 1,4-dicarbonilo que protege por la aegis de aromaticidad en el anillo de furano de 384 que se prepara fácilmente por litiación y luego alquilación de α-metilfurano.

La generación estereoselectiva del enlace C=C trans disustituido en 387 se logra mediante la modificación de Schlosser de la olefinación de Wittig. Así, el intermedio de β-oxidofosfonio 390, que es el producto principal de la adición de iluro 385 a un aldehído, se convierte en un intermedio epimérico de β-oxidofosfonio 393 por protonación de un iluro de β-óxido 392. Este carbanión se ve favorecido termodinámicamente sobre el carbanión epimérico 391. La eliminación de syn posterior (quizás a través de 2πs + 2πa cyclo- eliminación\(\ce{Ph3P=O}\) de un intermedio de betaína) entrega el alqueno trans estereoespecíficamente.

Aunque 387 no tiene funcionalidad polar en la posición 9, la posición 9 participa como electrófila y la posición 8 como nucleófila durante la ciclación polar a 388 en analogía con la anelación de 381 a 382 bis. La estereoselectividad de esta ciclación deriva de una preferencia estereoelectrónica por la adición antiperiplanar al enlace C=C en 387 y el control de aproximación estérica durante la formación del enlace entre las posiciones 8 y 14. La introducción estereoselectiva del metilo angular se logra mediante la generación del α-epóxido 389. Aquí SAC favorece el ataque β por\(\ce{Cl^+}\) sobre el enlace C=C. A esto le sigue la inversión configuracional estereoespecífica durante el desplazamiento intramolecular de S N 2. Finalmente, la migración 1,2-de metilo durante un reordenamiento de pinacol genera la configuración β-metilo requerida y una unión de anillo CD trans.
La estrategia más eficiente para la síntesis total de estrona es también la más convergente, uniendo una unidad 394 de anillo AB preformada con una unidad 380 de anillo D como en D. Esto, la síntesis Velluz 26, proporciona una fuente industrial de estrona que es más económica que la biosíntesis. La unión polar de 395 con 380 personifica el uso eficiente de la funcionalidad para facilitar la construcción esquelética. Curiosamente, 380 es un ácido carboxílico vinilólogo suficientemente ácido para protonar 395. El carbocatión resultante 396 se estabiliza por la funcionalidad relacionada con la diana en la posición 3, mientras que la nucleofilia en la posición 13 se estabiliza por la funcionalidad de oxígeno relacionada con la diana en la posición 17 en el enolato 397. La funcionalidad relacionada con el objetivo en la posición 3 también facilita la generación del enlace C-C 8-14 durante la ciclación catalizada por ácido de 400 a 382. Anteriormente se describió una ruta estereoselectiva de 382 a éter metílico de estrona (367).
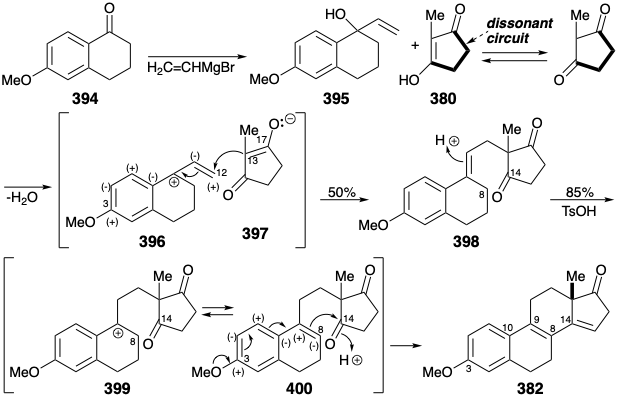
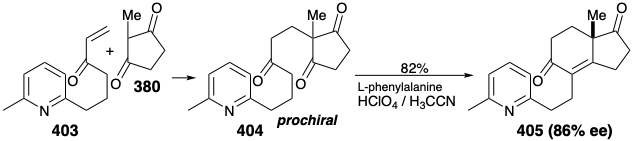
Una Síntesis Enantioselectiva. Todas las estrategias anteriores producen estrona racémica que debe resolverse para proporcionar el enantiómero natural. En 1975 se reportó una síntesis enantioselectiva de (+) -estrona natural. 27 El paso clave convierte la triona proquiral 404 en la diona enantioméricamente enriquecida 405 mediante una condensación de aldol enantioselectiva. Excepto por el uso de un anillo de piridina como precursor latente no nucleofílico de 1,5-dicarbonilo del anillo de estrona A, la estrategia topológica y polar es idéntica a la descrita anteriormente implicando la condensación de Michael de 379 con 380. En la presente síntesis, todos los carbonos requeridos para el objetivo final se unen en una condensación de Michael de 403 con 380. Después de la generación enantioselectiva crucial del anillo C, la unión del anillo CD trans requerida se establece mediante control de enfoque estérico durante la hidrogenación catalítica de un alcohol insaturado derivado de 405 por reducción selectiva con hidruro del carbonilo no conjugado más electrófilo. La finalización de las dos últimas conexiones esqueléticas requirió desenmascarar una matriz latente de 1,5-dicarbonilo por reducción de abedul de 406 seguido de hidrólisis catalizada por base del intermedio 1,4-dihidropiridina resultante y condensación aldólica. La hidrólisis del cetal en la posición 9 permite entonces el equilibrio del estereocentro C8 y la generación de la conexión esquelética final por otra condensación aldólica. La aromatización de 408 entrega estrona cruda. Una sola recristalización dio (+) -estrona ópticamente pura (13% de 380) así como estrona racémica (3% de 380) de las aguas madres.
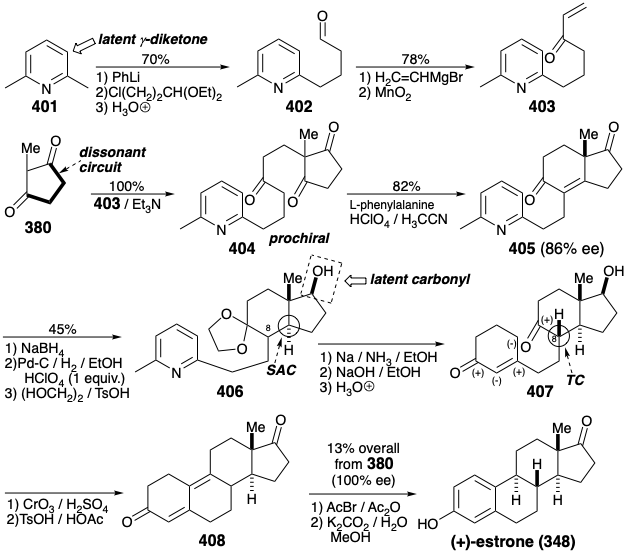
Síntesis de Estrona por Cicloadiciones. Dado que el anillo B de la estrona (348) contiene un enlace C=C en un anillo de seis miembros, es posible una síntesis de cicloadición térmica. Una reacción intramolecuar de Diels-Alder podría proporcionar estrona desde 409. Además, se puede emplear otro reordenamiento pericíclico para la síntesis de 409. Así, la matriz de 1,3-dieno en 409 puede generarse mediante reordenamiento electrocíclico de un ciclobuteno 410.
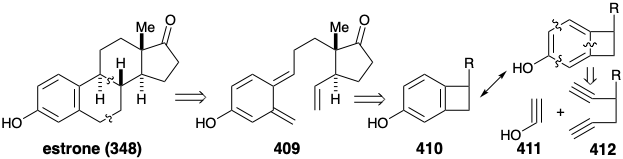
Una aplicación particularmente llamativa 28 de esta estrategia 29 genera un intermedio de benzociclobuteno 416 en una ciclotrimerización de alquino intramolecular catalizada por cobalto (I) que implica 1,5-diino 412 con un equivalente sintético 413 de la cetena enol 411. La trimerización es en realidad un proceso paso a paso que implica la adición oxidativa a cobalto (I) para generar un cobalto (III) ciclopentadieno 414. Diels Alder cicloadición de 413 con 414 luego produce 415 a partir de los cuales el catalizador de cobalto (I) es regenerado por una eliminación reductora que entrega 416.
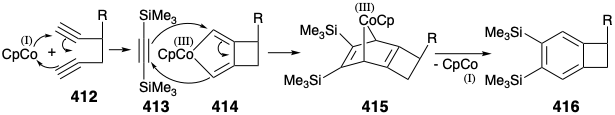
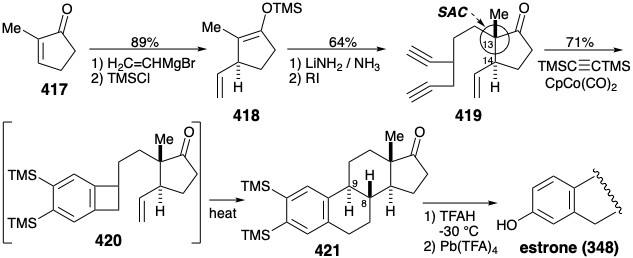
Las configuraciones relativas correctas en las posiciones 13 y 14 se establecieron mediante control de aproximación estérica durante la alquilación de un enolato que se produjo regioespecíficamente por 1,4-adición a 417 y regenerado a partir de 418. La cicloadición de 419 con bis (trimetilsilil) acetileno en presencia de ciclopentadienilcobalto dicarbonilo generó un intermedio benzociclobuteno 420. La generación promovida por calor del orto xileleno derivado seguida de cicloadición intramolecular proporcionó 421 en 71% de rendimiento global de 419. La generación estereoselectiva de las configuraciones correctas en las posiciones 8 y 9 surge de una preferencia por un estado de transición exo en la cicloadición de Diels-Alder. La congestión estérica desestabiliza el estado de transición endo alternativo que de otro modo sería favorecido por el solapamiento orbital secundario. La monoprotodesililación de 421 eliminó el grupo 2-sililo con una preferencia 9:1 sobre el grupo 3-sililo. La desililación oxidativa generó el 3-hidroxilo y liberó estrona en 24% de rendimiento global a partir de 2-metil-2-ciclopenten-1-ona (417) en siete etapas.
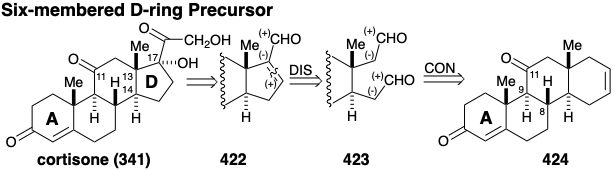
Síntesis Total de Cortisona. La estrategia para la primera síntesis total 30 de cortisona (341) se centró en el problema de ensamblar una transfusión entre los anillos C y D. El reto fue superar la preferencia termodinámica por una fusión cis entre un anillo de cinco y seis miembros. Dado que se favorece termodinámicamente una fusión trans entre dos anillos de seis miembros, una solución atractiva pareció ser crear un intermedio con un anillo D de seis miembros y contraer el anillo de seis a cinco miembros. Con este objetivo en mente, un grupo aldehído en la posición 17 en un precursor de ciclopenteno 422 debería proporcionar un punto de partida para construir la cadena lateral que se encuentra en esta posición en cortsona. La desconexión polar entre los carbonos 16 y 17 del anillo D sugiere un precursor de dialdehído 423 que proporcionaría 422 por condensación intramolecular de aldol. La conexión de los dos grupos aldehído reactivos en 423 sugiere un precursor de dialdehído latente 424 que contiene un anillo D transfusionado de seis miembros.
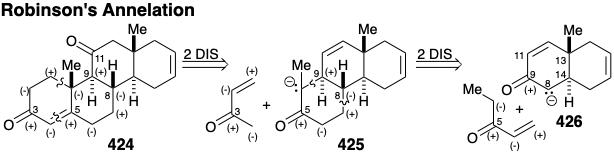
La desconexión de dos enlaces en el anillo A en la fusión del anillo resulta en una simplificación topológica importante. Los carbonos de fusión del anillo en el anillo A de 424 son átomos comunes, y la desconexión de estos dos enlaces, cada uno entre un átomo común y uno no común, elimina completamente el anillo A. Todos los circuitos entre los dos grupos carbonilo en 424 son disonantes. Por lo tanto, ambos carbonilos no pueden usarse simultáneamente para proporcionar activación polar para generar enlaces C-C. Más bien, se debe añadir funcionalidad adicional, por ejemplo un grupo carbonilo en la posición 5, a un precursor 425 para proporcionar activación electrófila en la posición 5 y activación nucleofílica en la posición 10 que puede explotarse junto con la funcionalidad relacionada con la diana en la posición 3 para ensamblar el A- anillo. Tal proceso de annelación de ciclohexenona polar, la “anelación Robinson”, se había ideado recientemente en el que una alquilvinil cetona sirve primero como electrófilo en la posición β del grupo vinilo y luego como nucleófilo en la posición α'. Así, se podría esperar que la condensación polar en dos etapas de 425 con metil vinil cetona proporcionara el anillo A en 424. También se podría usar un proceso similar para agregar el anillo B a un precursor bicíclico 426. Debido a que el grupo carbonilo en la posición 11 en la cortisona no puede proporcionar la reactividad polar requerida para la anelación descrita anteriormente, su introducción puede retrasarse hasta las últimas etapas de la síntesis.
La unidad de ciclohexeno en 427 sugiere una síntesis de cicloadición que implica una reacción de Diels-Alder entre un dienófilo precursor del anillo C y 1,3-butadieno. Sin embargo, la generación de una fusión de anillo trans requeriría un precursor de anillo C que contenga un enlace trans C=C severamente tensado en un anillo de seis miembros. Una estrategia más razonable sería generar la fusión de anillos trans favorecida termodinámicamente mediante la epimerización de un precursor cis fusionado que sería producido, a su vez, por una reacción de Diels-Alder de un enlace cis C=C en un dienófilo. El carbonilo en la posición 9 en 427 podría estar presente en forma latente en un precursor 428. Un grupo carbonilo en la posición 8 en 428 podría proporcionar la activación polar requerida para la epimerización de una fusión de anillo cis en 429 en una fusión de anillo trans. Este carbonilo también activaría el enlace C=C conjugado en 430 hacia la reacción de Diels-Alder con un dieno relativamente rico en electrones. Sin embargo, esta estrategia es fatalmente defectuosa porque se puede esperar que 430 aromatice por enolización para proporcionar 431. Para bloquear la aromatización y proporcionar activación adicional del dienófilo, se puede agregar un segundo carbonilo en la posición 12. Esto sugiere un dienófilo de p-quinona 434 que entregaría un aducto de fusión cis 433 por reacción de Diels Alder con 1,3-butadieno, y un intermedio transfusionado 432 por epimerización.
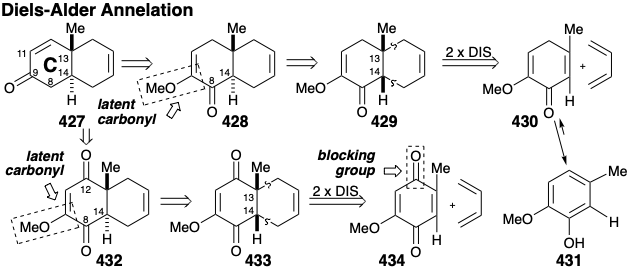
Se espera que el enlace C=C sustituido con metilo más deficiente en electrones en 434 sea un dienófilo más reactivo hacia 1,3-butadieno que el enlace C=C sustituido con metóxido más rico en electrones.
Después de haber proporcionado la activación para la epimerización en la posición 14 en 433, el sustituyente carbonilo en la posición 8 en 432 se elimina por reducción a un hidroxilo, acetilación y escisión reductora. Simultáneamente, el grupo carbonilo bloqueante en la posición 12 se elimina por reducción y deshidratación proporcionando 427. La activación nucleofílica en la posición 8 se potencia luego mediante la adición de un grupo formilo que existe en la forma enol en 435. La anelación de Robinson con etil vinil cetona (EVK) crea entonces el anillo B en 437. Así, después de facilitar la alquilación de Michael, el grupo formilo activador se elimina mediante una condensación retro de Claisen tras el tratamiento con\(\ce{KOH}\). Simultáneamente,\(\ce{KOH}\) cataliza una condensación intramolecular aldólica en 436 generando el anillo B en 437.
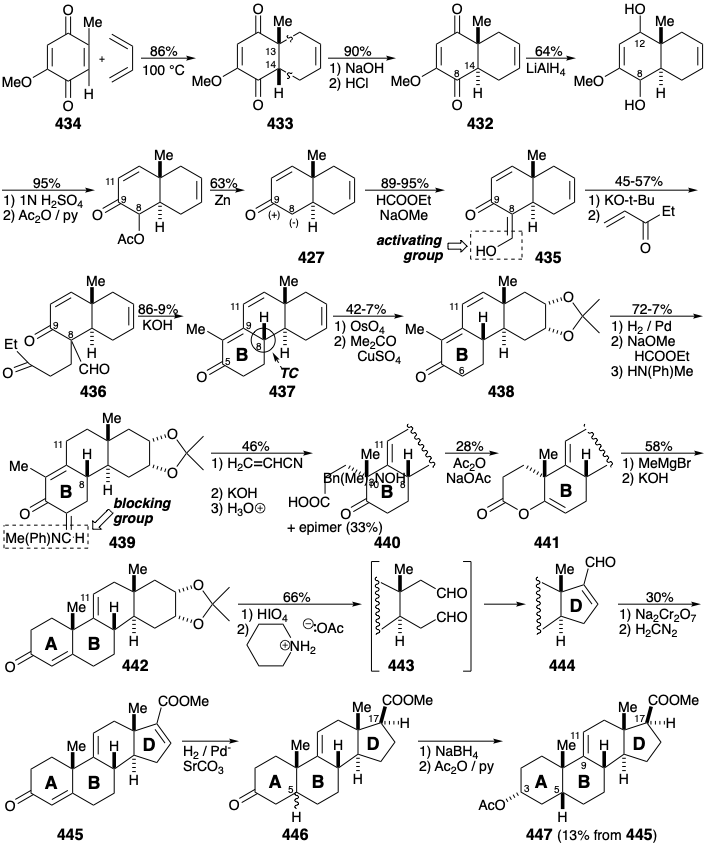
La estereoselectividad de esta anelación puede atribuirse a una preferencia termodinámica por la configuración requerida en la posición 8 que es epimerizable debido a la conjugación con el grupo carbonilo en la posición 5. La adición dioxidativa selectiva al enlace C=C no conjugado en el trieno 437 es factible porque el electrófilo\(\ce{OsO4}\) es menos reactivo hacia los enlaces C=C conjugados deficientes en electrones. La saturación selectiva del enlace C=C estéricamente menos gravado y menos sustituido en 438 es seguida por la instalación de un derivado enamina de un sustituyente formilo como grupo bloqueante en la posición 6 en 439. 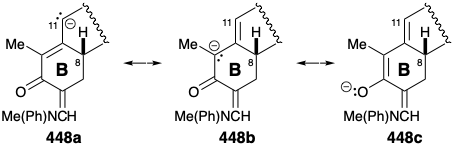 Destacan las aplicaciones contrastantes de los sustituyentes formilo en 436 y 439. De los tres hidrógenos ácidos restantes en 439, la abstracción de protones de la posición 11 está menos gravada estéricamente. El nucleófilo dienolato ambiente resultante, como se esperaba, es selectivamente Michael alquilado α al grupo carbonilo debido a una mayor densidad electrónica en el centro en comparación con el átomo de carbono terminal de la matriz 1-oxa-pentadienilo en el carbanión intermedio. En otras palabras, la forma de resonancia 448b es más importante que la 448a de 448c.
Destacan las aplicaciones contrastantes de los sustituyentes formilo en 436 y 439. De los tres hidrógenos ácidos restantes en 439, la abstracción de protones de la posición 11 está menos gravada estéricamente. El nucleófilo dienolato ambiente resultante, como se esperaba, es selectivamente Michael alquilado α al grupo carbonilo debido a una mayor densidad electrónica en el centro en comparación con el átomo de carbono terminal de la matriz 1-oxa-pentadienilo en el carbanión intermedio. En otras palabras, la forma de resonancia 448b es más importante que la 448a de 448c.
Desafortunadamente, la alquilación de 448 es no estereoselectiva produciendo ácido carboxílico 440 y un epímero después de la hidrólisis de un nitrilo intermedio. El carbono final requerido para el anillo A de la cortisona debía ser proporcionado por\(\ce{MeMgBr}\). Sin embargo, para evitar la adición a la cetona carbonilo en 440, este último grupo se enmascaró como una enol lactona en 441. La metil cetona producida por adición de\(\ce{MeMgBr}\) a 441 suministró 442 por condensación intramolecular aldólica. El bajo rendimiento para la conversión 440 a 442 se mejoraría, sin duda, por métodos modernos. Así, una reacción quimioselectiva del cloruro de ácido de 440 con\(\ce{LiMe2Cu}\) proporcionaría un alto rendimiento de la metil cetona correspondiente.
La contracción del anillo D se logró mediante escisión oxidativa al dialdehído 443 que dio 444 por una condensación aldólica notablemente regioselectiva. La saturación de los dos enlaces C=C menos estéricamente gravados en el éster derivado 445 dio una mezcla de epímeros 446 (en la posición 5) separación de los cuales se logró mejor después de la conversión al derivado 3-hidroxi que luego proporcionó 447 por acetilación.
En este punto la síntesis total se cruzó con “extensas investigaciones previas de muchos grupos sobre la síntesis parcial, a partir de fuentes naturales, de cortisona”. Así, la introducción del grupo 11 carbonilo se logró mediante epoxidación del alqueno 447, hidrólisis del epóxido a triol 449 y oxidación a diona hemicetal 450. El reemplazo nucleofílico del oxígeno en la posición 9 por Br procedió con reordenamiento alílico al tratamiento de 450 con\(\ce{HBr}\). La generación selectiva de una fusión de anillo trans A-B y la configuración requerida en la posición 9 en 451 es consecuencia del control termodinámico. La desbromación reductiva de la bromocetona 451 seguida de la reducción a diol, la acetilación selectiva del hidroxilo menos estéricamente gravado en la posición 3 y la oxidación liberaron el 11-ceto-esteroide 452.

El ensamblaje de la cadena lateral de cortisona disonante se realizó mediante condensación polar del cloruro de ácido de 452 con un bloque de construcción disonante, diazometano y reacción de una diazocetona intermedia con ácido acético. Saponificación del diacetato resultante 453 y reacetilación selectiva del hidroxilo primario entregado 454. La introducción de un grupo hidroxilo en la posición 17 se logró luego mediante la adición 1,2-dioxidativa a 455. El control de aproximación estérica favoreció la configuración requerida en la posición 17. Finalmente, la introducción de insaturación entre los carbonos 4 y 5 se logró mediante bromación seguida de deshidrobromación de 456. La deshidrobromación de α-bromocetonas puede complicarse por la sustracción de protones α' que conduce a una reacción de Favorskii a través de la formación de una ciclopropanona. El método suave de Mattox-Kendall evita este escollo porque las α-bromo hidrazonas eliminan fácilmente el bromuro para generar hidrazonas intermedias α, β-insaturadas, por ejemplo 457. El ácido pirúvico afecta la eliminación de la hidrazina mientras que la hidrólisis del acetato de la cadena lateral se produjo tras el tratamiento con la\(\ce{HCl}\) finalización de la primera síntesis total de cortisona (341) por R. B. Woodward en 1952.

Como fuente práctica de suministro para aplicaciones medicinales, la síntesis de Woodward no podría competir con síntesis parciales de otros esteroides naturales que están fácilmente disponibles en las plantas. Un déficit importante de la síntesis total es la generación de un producto racémico. El enantiómero natural estuvo disponible por resolución del intermedio 445, pero al menos la mitad de este precioso intermedio avanzado se descartó, es decir, el enantiómero equivocado. Aunque la biosíntesis de cortisona es circuitosa, produce solo un enantiómero debido a una epoxidación completamente enantioselectiva de escualeno.
Una estrategia enantioselectiva para cortisona. El logro de una síntesis total industrialmente viable de cortisona dependió del desarrollo, en 1966, de una estrategia enantioselectiva. 31 Curiosamente, la estrategia evolucionó a partir de métodos e intermedios desarrollados durante una síntesis de cortisona que emplea metoxitetralona (394) como precursor del anillo BC 32 en contraste con la síntesis de estrona Velluz mencionada anteriormente (ver sección 5.3) que utilizó esta material de partida para el grupo de anillo AB. Pero el desarrollo clave en la evolución de una estrategia enantioselectiva fue la concepción de un material de partida proquiral y una ruta para su elaboración en un esteroide. 33 Así, una extensa investigación sobre la síntesis parcial de cortisona a partir de 17-cetoesteroides de origen natural había establecido métodos que permiten la elaboración de la cadena lateral de cortisona a partir de precursores como 458.
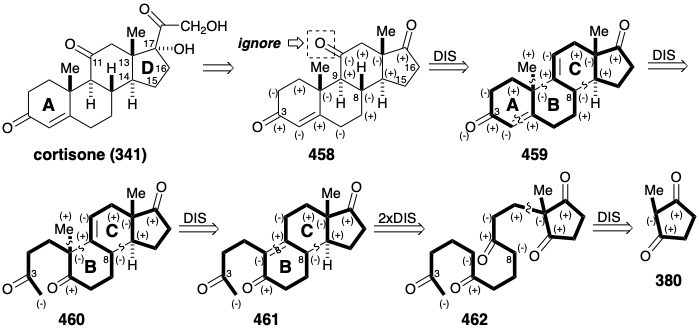
El análisis polar de esta subdiana, ignorando el grupo 11-keto, revela que todos los circuitos entre la funcionalidad oxígeno en los anillos A y D son consonantes excepto aquellos que involucran los carbonos 15 y 16. La introducción del grupo 11-ceto por adición a un enlace C=C del anillo C en un precursor 459 está precedida por la funcionalización similar de 447 (ver sección 5.4) en la síntesis de Woodward.
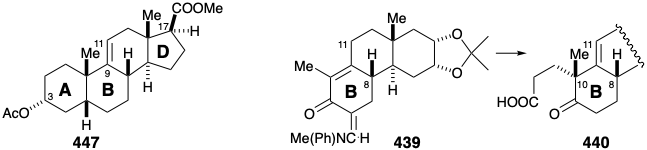
La desconexión polar del enlace 4,5-C=C en 459 sugiere una condensación intramolecular aldólica de 460 para generar el anillo A. En la síntesis de Woodward, se generó un intermedio similar, es decir, 440, por alquilación de 439. La metilación del análogo 461 de 439 podría proporcionar 460 de manera similar. Otras desconexiones polares de los anillos B y C sugieren un precursor monocíclico 462 que es aquiral. Vimos un intermedio similar en la síntesis enantioselectiva de estrona de Danishefsky. Así, 404 (ver arriba) contiene dos de los grupos carbonilo de 462 en forma latente. En la síntesis de estrona, la asimetría se introdujo mediante una condensación aldólica enantioselectiva de 404 para dar 405. También, en esa síntesis, 404 se preparó a partir de un precursor 380 de anillo D intacto por adición de Michael a la enona 403.
Una década antes se empleó una estrategia similar para preparar un precursor proquiral análogo para la cortisona. Sin embargo, como veremos, se utilizó una transformación enantioselectiva diferente del precursor proquiral 467 (ver más adelante) para introducir asimetría en esta síntesis de cortisona por Velluz.
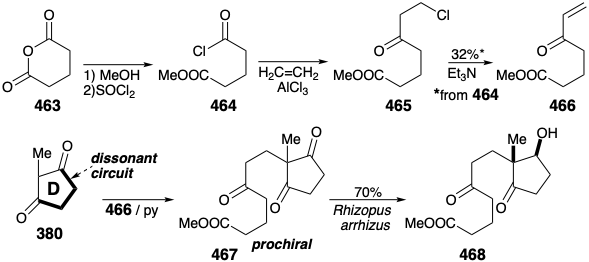
Un material de partida simétrico 463 se convierte en el aceptor Michael 466 a través de la acilación Friedel-Crafts de etileno con el cloruro de ácido 464 y la deshidrocloración del intermedio β-cloro cetona 465. Aunque el rendimiento de 466 es pobre, está fácilmente disponible en cantidades de kilogramos a partir de materiales de partida económicos. La condensación de 466 con el precursor 380 intacto del anillo D entrega 467. La reducción microbiológica enantioselectiva proporciona el producto de mono reducción ópticamente puro 468 con buen rendimiento como un solo diastereómero.
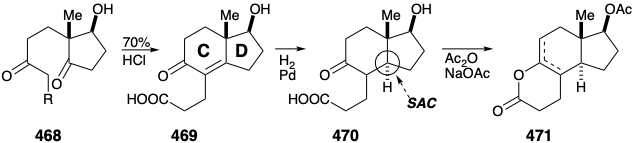
La condensación intramolecular de aldol catalizada por ácido crea entonces el anillo C en 469. La saturación estereoselectiva del enlace C=C en 470 crea la fusión de anillo trans CD requerida, sin necesidad del largo proceso de contracción del anillo de la construcción Woodward de la fusión de anillos trans CD. Para lograr la conexión polar selectiva en el carbono carboxilo en 470 de un fragmento nucleofílico que contiene los carbonos restantes requeridos para los anillos A y B, se debe enmascarar la cetona carbonilo. Esto se logra intramolecularmente mediante esterificación enólica para dar 471.
El reactivo de Grignard 472 reacciona con 471 para suministrar 473 que se cicla por condensación intramolecular aldólica para proporcionar el anillo B en 474. La configuración correcta en la posición 8 está asegurada por el control termodinámico debido a que este centro está conjugado con el carbonilo en la posición 5. Sin embargo, la abstracción de protones de 474 ocurre principalmente en la posición 11 conduciendo a la metilación angular a 475 completamente estereoselectivamente con la configuración requerida para la cortisona. La estereoselectividad de esta alquilación contrasta fuertemente con la falta de estereoselectividad observada en la conversión correspondiente de 384 a 385 en la síntesis de Woodward. El comportamiento estereoquímico contrastante de estas dos alquilaciones es consecuencia de diferencias mecanicistas. Así, la alquilación de 439 es una reacción de Michael reversible, controlada termodinámicamente, mientras que la metilación de 474 es un proceso controlado cinéticamente. La metilación axial de la cara β del enolato se prefiere estereoelectrónicamente porque conduce directamente al conformador silla de 475.
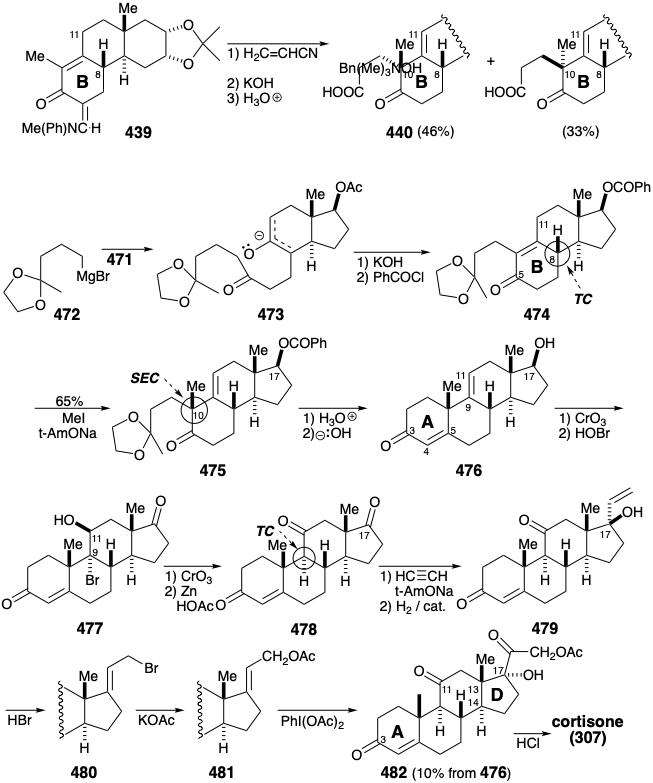
La generación del anillo A se logra entonces con una tercera condensación intramolecular aldólica que proporciona 476 después de la hidrólisis del benzoato 17. La oxidación del hidroxilo 17, y la introducción de oxígeno en la posición 11 por adición de\(\ce{HOBr}\) proporciona 477 que, tras la oxidación y desbromación reductiva, entrega 478 estereoselectivamente con la configuración termodinámicamente preferida en la posición 9 que es α a la carbontilo en la posición 11. Finalmente, la elaboración de la cadena lateral de cortisona se logró por etinilación del carbonilo 17 no conjugado más electrófilo. La hidrogenación catalítica proporcionó entonces el alcohol alílico terciario 4 7 9. La bromodehidroxilación de 4 7 9 ocurrió con reordenamiento alílico, presumiblemente a través de un intermedio catiónico alílico, mientras que la sustitución de\(\ce{Br}\) en 480 con\(\ce{OAc}\) procedió por una sustitución directa de S N 2. El agente oxidante inusual, feniliodosodiacetato y una cantidad catalítica de\(\ce{OsO4}\), convirtió el enlace C=C más nucleófilo en 481 directamente en la α hidroxicetona 482.