4.6: Biosíntesis y Síntesis Total de Diterpenos - Spatol
- Page ID
- 70296

Biosíntesis
En cuanto a todos los productos naturales, existía una estrategia sintética exitosa para el espatol antes de cualquier esfuerzo humano. Siempre es interesante examinar la estrategia de la Naturaleza porque un enfoque análogo, una estrategia biomimética (imitando a la Naturaleza), puede ser efectiva en el laboratorio. Así, los diterpenos son compuestos C 20 derivados biogenéticamente de E, E, E-geranilgeranil-PP (192) o sus isómeros geométricos.
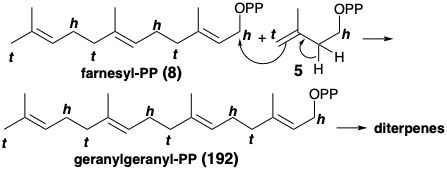
Estos tetrámeros acíclicos de Δ 3-isopentenil-PP (5) surgen de la reacción de 5 con un trímero como E, E-farnesil-PP (8). La posterior adición electrófila intramolecular del pirofosfato alílico a los enlaces C=C trisustituidos puede conducir a diversos carbocationes mono y multicíclicos como 193. Otra ruta general hacia los electrófilos carbocatiónicos implica la protonación de enlaces C=C. Una vía hipotética para la biosíntesis de espatol (196) implica la protonación de un enlace C=C y la adición electrófila intramolecual del carbocatión resultante para producir un ciclobutano (195) por pérdida de protones de 194. Esta hipótesis deriva el apoyo de la ocurrencia natural de 195. 14 Se presume que la funcionalidad oxígeno en 196 surge del metabolismo oxidativo de 195 por los organismos marinos que producen este dieno tricíclico y una amplia variedad de metabolitos oxigenados con el esqueleto de carbono de espatana.
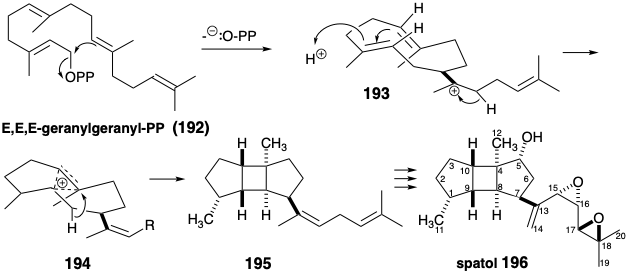
Análisis Topológico de Spatol
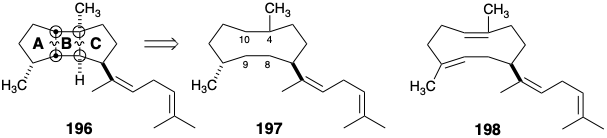
El análisis retrosintético topológico ex post facto de la estrategia biosintética revela una característica importante. El núcleo triciclo [5.3.0.0 2,6] decano de los diterpenos de espatano incorpora 4 átomos comunes (rodeados en 196), los cuatro átomos del anillo B. La estrategia biosintética se beneficia de la poderosa simplificación topológica que se deriva de la eliminación de enlaces entre dos conjuntos de átomos comunes, 4-8 y 9-10. Esto sugiere un sintón topológico monocíclico 197. Para este sintón, un equivalente sintético, 198, es sugerido por nuestra hipótesis biogenética.
Otro equivalente sintético, 199, se sugiere por la posibilidad de una cicloadición intramolecular 2π + 2π. Una síntesis expedita 15a del sesquiterpeno b-borboneno (2 0 1) explota la fotocicloadición intramolecular de germacreno D (200) un intermedio análogo al 199. La evidencia UV (λ max = 259 mm, ε = 4500 en n-hexano) indica una interacción transanular significativa entre los dos enlaces endocíclicos C=C en el sesquiterpeno 200. Así, 200 probablemente prefiere una conformación en la que los dos enlaces C=C endocíclicos están situados paralelos y cara a cara entre sí, y el sustituyente isopropilo ocupa la configuración ecuatorial menos estéricamente impedida. El control termodinámico de la conformación de 200 asegura la configuración adecuada en el carbono portador de isopropilo mientras que el control estereoelectrónico (syn periplanar = adición suprafacial) asegura las configuraciones cis, anti, cis correctas en los estereocentros de ciclobutano. Una fotociclación análoga para generar un precursor 202 para el espatol no es favorable ya que un sustituyente R que va a convertirse en la cadena lateral debe ocupar una posición axial más estéricamente gravada como en 202a en lugar de la conformación más termodinámicamente favorable 202 e.
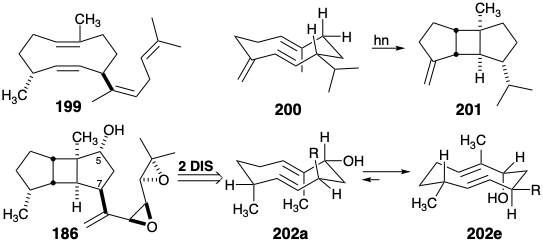
Una estrategia topológica y estereoquímica
También se ha aprovechado para su síntesis una disección topológica diferente del núcleo cis-anti-cis-triciclo [5.3.0.0 2,6] decano del β-bourboneno (201). Así, la eliminación de dos enlaces entre pares de átomos comunes puede generar dos precursores de ciclopenteno, 203 y 204, que podrían estar unidos por una fotocicloadición de 2πs + 2πs (ver sección 3.3). De hecho, la irradiación UV de 2-ciclopenten-1-ona con 204 da como resultado una fotocicloadición que es orientacionalmente no selectiva, produciendo una mezcla 1:1 de isómeros estructurales 205 y 206. 15b Sin embargo, la cicloadición es favorablemente estereoselectiva debido a una preferencia controlada por el enfoque estérico para la cicloadición a la cara del anillo de ciclopenteno opuesta al sustituyente isopropilo. Esta estereoselectividad resta utilidad a una síntesis similar para el spatol porque la cadena lateral del diepóxido alílico en spatol (196) es cis al ciclobutano en lugar de trans como lo es el grupo isopropilo en 201 o 205.
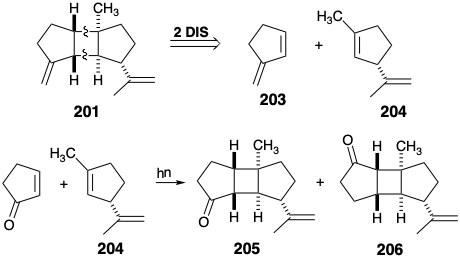
Así, se puede esperar que la cadena lateral del diepóxido alílico o su precursor en un intermedio de ciclopenteno 207 favorezca la estereoselectividad incorrecta en una fotocicloadición con 2-ciclopenten-1-ona, es decir, favoreciendo 208 o 209 en lugar del aducto deseado 210 o su isómero estructural 211.
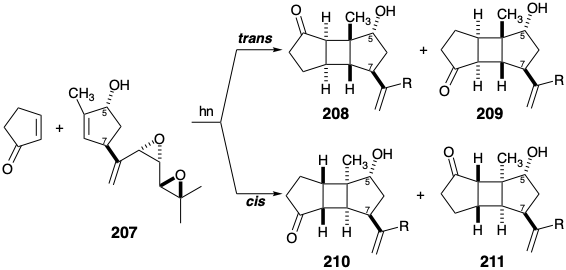
Una estrategia para superar esta deficiencia de fotocicloadiciones de ciclopenteno para la síntesis total de espatol utiliza un puente temporal para proteger una cara de un anillo de ciclopenteno para impedir la adición a esa cara. 16 Así, dicho puente se puede proporcionar uniendo el grupo hidroxilo en la posición 5 con un grupo carboximetilo que también sirve como progenitor de la cadena lateral en la posición 7 como en la lactona 213. Además, la lactona puede derivarse de un precursor latente, la cetona 214, mediante una oxidación de Baeyer-Villiger. La doble desconexión de 214 por una cicloeliminación sugiere fotocicloadición de una norbornenona 215 con ciclopent-2-en-1-ona. Así, en 214 un puente oxoetano temporal protege la cara α del incipiente anillo C imponiendo la cicloadición estereoselectiva del precursor del anillo A ciclopent-2-en-1-ona trans al incipiente grupo 5-hidroxilo.
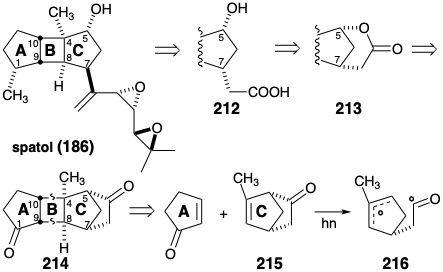
Esta estrategia tiene un defecto obvio. Se esperaba que la cetona homoalílica bicíclica colada 215 experimentara fácilmente escisión fotoinducida a dirradical 216. Sin embargo, el enmascaramiento del carbonilo en 215 evitaría este problema y facilitaría la diferenciación entre los dos grupos carbonilo en el fotocicloaducto 214. Además, la configuración en la posición 7 en 212 tendría que ser invertida para proporcionar la configuración requerida en este estereocentro en espatol. Una síntesis de 6-metilbiciclo [2.2.1] hept-5-eno-2-ona (215) a partir de acetato de vinilo y metil-1,5-ciclopentadieno es posible a través de una reacción de Diels-Alder de estos materiales de partida. Aunque la reacción produce una mezcla de isómeros estructurales, la saponificación seguida de oxidación da una mezcla de cetonas isoméricas de las cuales 215 pueden aislarse por destilación.
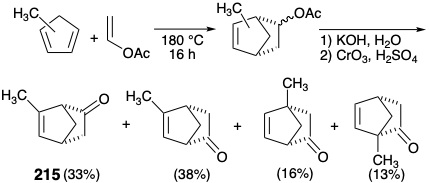
Enmascarar una cetona sensible
El enmascaramiento del grupo carbonilo en 215 resultó inesperadamente difícil. Solo se obtuvo un rendimiento moderado del cetal de etileno 217 por cetalización catalizada por ácido de 215 bajo condiciones que dan un excelente rendimiento de cetal a partir del análogo 6-no substituido, biciclo- [2.2.1] hept-5-en-2-ona, debido a una fragmentación competitiva a 218. La proclividad de 215 hacia esta fragmentación surge indudablemente de la estabilidad relativa del carbocatión terciario 220 y el alivio de la cepa anular que asiste a la conversión de 219 a 220.
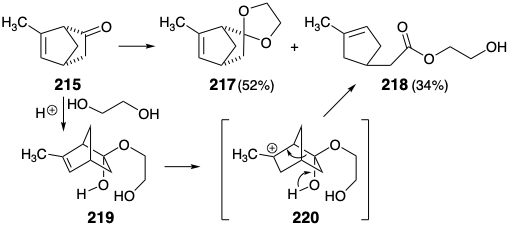
Se desarrolló una elección inusual para el grupo enmascarante durante la búsqueda de un grupo que pudiera introducirse en condiciones no ácidas. Así, la cetona 215 se convierte cuantitativamente en una mezcla epimérica de cianohidrina silil éteres 221 por reacción con cianuro de trimetilsililo. Si bien el uso de un grupo enmascarante asimétrico puede parecer imprudente ya que esto conduce a mezclas epiméricas de varios intermedios, este es un precio pequeño a pagar por las características que de otro modo serían ideales del grupo enmascarante de cianhidrina silil éter. Así, la fotocicloadición con ciclopent-2-en-1-ona liberó dos aductos epiméricos 222x y 222n con alta selectividad estéreo (cis, anti, cis, fusiones de anillo y exo adición al biciclohept [2.1.1] eno) y orientacional (ciclopentano carbonilo alejado del grupo metilo de cabeza de puente). Por suerte, el aducto principal 222x cristalizó de la mezcla de fotorreacción junto con el dímero de ciclopentenona del que se separó fácilmente por trituración con hexano caliente dejando atrás el dímero puro. 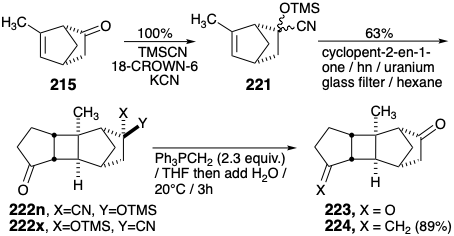 Luego se obtuvo 222x puro con 51% de rendimiento, basado en 2 2 1, por elución del producto parcialmente purificado a través de una columna de gel de sílice con acetato de etilo-hexano. La cromatografía en columna del fotoproducto soluble en hexano proporcionó una fracción a partir de la cual el cicloaducto 222n menor casi puro cristalizó junto con un poco de 222x. Esta mezcla es adecuada para la olefinación de Wittig para producir metilidenocetona 224, vide infra. El rendimiento combinado aislado de 222x más 222n supera 60%. Se obtuvo una muestra de 222n puro por HPLC. La relación epimérica entre 222x y 222n se demostró mediante la producción de la misma dicetona 223 tras la hidrólisis del grupo enmascarante de cianhidrina silil éter y también por la producción de la misma metilidencetona 224 tras la reacción con metilenetrifenilfosforano seguido de hidrólisis del éter silílico de cianohidrina. La conversión de 222 en 224 se realizó como un procedimiento de un solo recipiente proporcionando 224 puro en 89% de rendimiento global. La utilidad del grupo enmascarante de cianohidrina silil éter en las transformaciones anteriores es notable. Se introduce en condiciones de reacción neutras suaves. Es suficientemente robusto para sobrevivir a la irradiación UV, la cromatografía sobre gel de sílice y la olefinación de Wittig; pero se convierte fácilmente en un grupo carbonilo por la base acuosa generada tras la adición de agua a la mezcla de reacción de Wittig.
Luego se obtuvo 222x puro con 51% de rendimiento, basado en 2 2 1, por elución del producto parcialmente purificado a través de una columna de gel de sílice con acetato de etilo-hexano. La cromatografía en columna del fotoproducto soluble en hexano proporcionó una fracción a partir de la cual el cicloaducto 222n menor casi puro cristalizó junto con un poco de 222x. Esta mezcla es adecuada para la olefinación de Wittig para producir metilidenocetona 224, vide infra. El rendimiento combinado aislado de 222x más 222n supera 60%. Se obtuvo una muestra de 222n puro por HPLC. La relación epimérica entre 222x y 222n se demostró mediante la producción de la misma dicetona 223 tras la hidrólisis del grupo enmascarante de cianhidrina silil éter y también por la producción de la misma metilidencetona 224 tras la reacción con metilenetrifenilfosforano seguido de hidrólisis del éter silílico de cianohidrina. La conversión de 222 en 224 se realizó como un procedimiento de un solo recipiente proporcionando 224 puro en 89% de rendimiento global. La utilidad del grupo enmascarante de cianohidrina silil éter en las transformaciones anteriores es notable. Se introduce en condiciones de reacción neutras suaves. Es suficientemente robusto para sobrevivir a la irradiación UV, la cromatografía sobre gel de sílice y la olefinación de Wittig; pero se convierte fácilmente en un grupo carbonilo por la base acuosa generada tras la adición de agua a la mezcla de reacción de Wittig.
Amplificador SAC
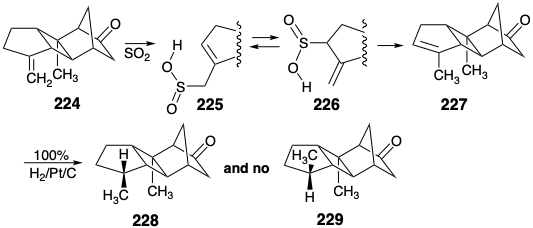
La hidrogenación catalítica de 224 favoreció el epímero 228 requerido sobre el subproducto inútil 229 por 10:1 debido al control de aproximación estérica por el grupo metilo en 224 que protege la cara α del anillo A. Sin embargo, la separación de 228 de la mezcla no se pudo lograr por ningún método excepto la cristalización fraccionada, y esto solo permitió el aislamiento del epímero deseado con un rendimiento justo. Para eludir este problema de separación, 224 se isomerizó al alqueno endocíclico 227 por\(\ce{SO2}\). Esta isomerización limpia y cuantitativa implica presumiblemente una adición de\(\ce{SO2}\) hasta 224 produciendo 225. Posterior [1.3] reordenamiento sigmatropico de azufre proporciona 226 que experimenta fragmentación retro ene entregando 227. La hidrogenación catalítica de 227 entrega 228 limpia y cuantitativamente. Al parecer, la proximidad más cercana del enlace C=C enodíclico al grupo metilo en 227 que el C=C exocíclico al grupo metilo en 224 da como resultado un mayor impedimento estérico para el suministro de hidrógeno α en 227 que en 224.
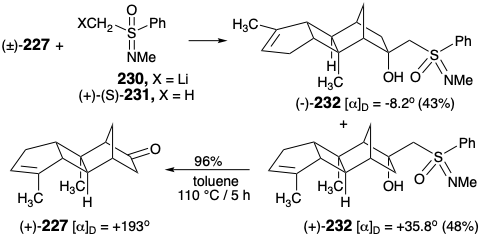
Resolución
La resolución eficiente y virtualmente cuantitativa de la cetona 227 se logró fácilmente mediante cromatografía flash y cristalización del aducto 1,2-con litiosulfoximina quiral 230. La eliminación retro ene del diastereómero dextrorrotatorio menos soluble (+) - 232 liberó cetona (+) - 227 que se correlacionó con (+) -spatol por conversión a un producto de degradación a partir de espatol natural (vide infra). La sulfoximina (+) - (S) - 231 se recuperó con 96% de rendimiento.
Un puente temporal durante la entrega de hidruro
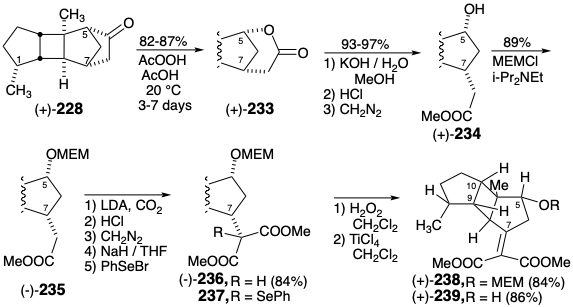
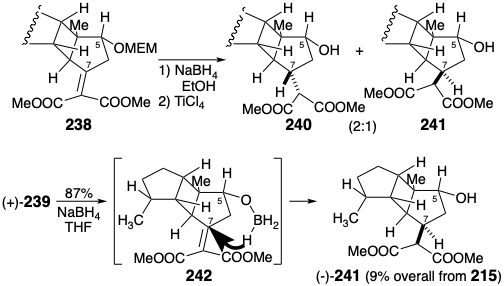
La hidrogenación catalítica de (+) - 227 proporcionó cetona (+) - 228. Luego se abordó la introducción de oxígeno en la posición 5, la escisión del puente temporal y la inversión de la configuración en la posición 7. Así, la oxidación de Baeyer-Villiger para dar lactona (+) - 233, saponificación y metilación del ácido resultante proporcionó alcohol (+) - 234. El enmascaramiento del 5-hidroxilo proporcionó (-) - 235 que se carboxiló para dar éster malónico (-) - 236. La epimerización en la posición 7 se inició mediante selenenilación seguida de deselenilación oxidativa del 237 resultante para entregar el éster alquiliden-malónico 238. La reducción de 238\(\ce{NaBH4}\) seguido de la eliminación del grupo protector MEM\(\ce{TiCl4}\) proporcionó una mezcla 2:1 respectivamente del éster cis-hidroxi malónico 240 y su epímero C-7, el éster malónico transhidroxilado deseado 241. . Este resultado decepcionante sugirió que el sustituyente 2-metoxietoximetoxi (OMEM) en la posición 5 dificulta estéricamente el suministro de hidruro a la cara α- del enlace C=C en 238.
Se encontró que un grupo hidroxilo remoto fomentaba el suministro de hidruro de syn pseudointramolecular a través de un alcoxiborohidruro intermedio 242. Así, el tratamiento del derivado de hidroxialquilideno malonato (+) - 239\(\ce{NaBH4}\) con el éster transhidroxi malónico deseado (-) - 241 completamente estereoselectivamente. El rendimiento global fue de 9% del precursor de anillo C 215 en 21 etapas incluyendo la resolución.
Síntesis enantioespecífica con un auxiliar quiral
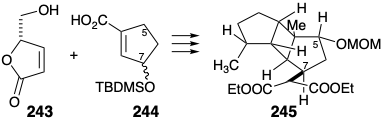
Se diseñó una síntesis diferente 18 de un éster triciclodecilmalónico homoquiral intermedio 245 (un análogo de 241) con un enfoque en explotar butenólido 243 como auxiliar quiral para establecer la configuración absoluta correcta durante la generación del B- anillo por una fotocicloadición 2π + 2π con precursor de anillo A 244. El butenólido homoquiral 243 está fácilmente disponible a partir del ácido L-glutámico. Un sustituyente alílico de oxígeno en 244 proporciona un punto de unión para la cadena lateral del éster malónico y activación para la introducción de oxígeno en la posición 5.
En un formato retrosintético, la estrategia contempla la unión del último éster malónico mediante una alquilación estereoespecífica S N 2 con 246. Dado que el auxiliar quiral 243 no proporciona el anillo de ciclopentano requerido para el anillo A de 245, este anillo tendrá que generarse después de la fotocicloadición de 250.
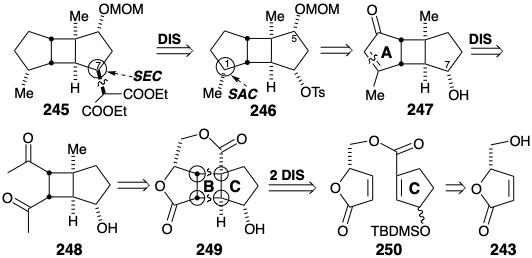
El anillo A podría ser creado por una condensación intramolecular aldólica de un precursor de bis metilcetona 248. Este paso es potencialmente defectuoso porque es posible una condensación alternativa indeseable de aldol. Sin embargo, un excelente precedente es proporcionado por un paso similar en una síntesis total del sesquiterpeno α- bourboneno (F). Así, la condensación intramolecular aldólica de A suministra ciclopentenona E y ninguna de la ciclopentenona C isomérica. Aparentemente, la ciclación al producto B de condensación aldólica se ve desfavorecida por el impedimento estérico por el sustituyente metilo angular en el producto alternativo D de condensación aldólica. El control de aproximación estérica (SAC) debería favorecer el β-suministro de hidrógeno durante la reducción de 247 para dar la configuración requerida en la posición 1 en 246. La orientación adecuada durante la generación del anillo B se puede asegurar mediante un puente temporal, un éster, entre 243 y el precursor 244 del anillo C.
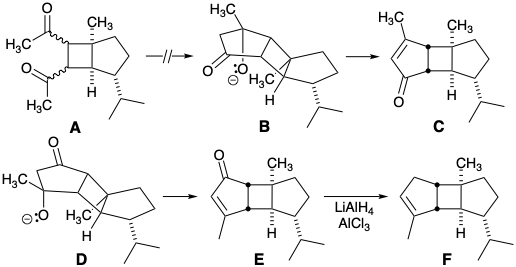
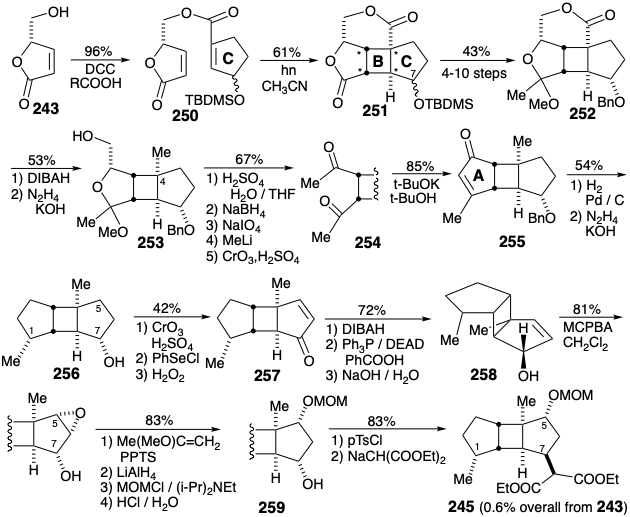
La fotocicloadición intramolecular del éster 250 del auxiliar quiral 243 suministró ciclobutano 251. La adición de un grupo metilo, el carbono necesario para completar el anillo A requirió 4-10 etapas dependiendo de la configuración del sustituyente C-7 en 251. La reducción del éster restante en 252 proporcionó entonces el grupo metilo en la posición 4 en 253 el cual había sido funcionalizado únicamente para permitir la construcción del puente temporal en 250. La manipulación del grupo funcional proporcionó entonces la diona 254 que se sometió a condensación aldólica completamente selectiva proporcionando 255. La hidrogenación estereoselectiva de 255 creó el estereocentro en la posición 1 y eliminó el grupo protector bencilo. La reducción de Wolff-Kishner de la cetona saturada resultante entregó 256. La introducción de los sustituyentes 5-hidroxilo y 7-malónico éster requirió entonces oxidación a 257, reducción e inversión de Mitsunobu para dar 258, epoxidación estereoselectiva seguida de reducción de hidruro, protección y desprotección para suministrar 259 y nucleófilo sustitución que proporcionó el éster malónico 245 en 0.6% de rendimiento global de 243 en 29-35 etapas.
Estrategias convergentes y lineales
El rendimiento global del éster malónico homoquiral (-) - 241 a partir del racémico (±) - 215 en la primera síntesis fue de 9%, más de un orden de magnitud superior al rendimiento total de 0.6% del éster malónico homoquiral 245 de homoquiral 243 en la segunda síntesis. El éxito de la primera síntesis, que se basa en la resolución para introducir asimetría, es especialmente notable porque la resolución es inherentemente ineficiente: proporciona en el mejor de los casos como 50% de rendimiento del enantiómero correcto. Dos factores disminuyen la penalización por usar resolución. En primer lugar, la resolución se realiza muy temprano en la primera síntesis y, por lo tanto, se minimiza el esfuerzo desperdiciado al desechar la mitad del producto racémico. Segundo, el método de sulfoximina de Johnson es espectacularmente efectivo. Además, las ventajas del plan inteligente para explotar el auxiliar quiral fácilmente disponible 243 para introducir asimetría en la segunda síntesis no pueden superar la penalización que surge de la ausencia de un grupo metilo en la posición 4 o un grupo hidroxilo en la posición 5, y la falta de un anillo A en el fotocicloaducto 251 de 243. La primera síntesis es más convergente que la segunda. Así, se construyen dos grandes fragmentos que contienen la mayoría o la totalidad de los átomos esqueléticos y la funcionalidad de la diana y estos fragmentos se unen luego. Tal enfoque tiene varias ventajas sobre una síntesis lineal, es decir, aquella en la que la molécula se construye uniendo secuencialmente muchos fragmentos pequeños o introduciendo funcionalidad después de que se complete la construcción esquelética. Una síntesis convergente es más eficiente según se mide por el rendimiento global. Si el rendimiento promedio de una síntesis de n etapas es de ψ%, entonces el rendimiento global será de 100 (ψ/100) n%. Una síntesis lineal de 21 etapas con un rendimiento promedio de 95% tendrá un rendimiento general de 34%, o un 11% general con un rendimiento promedio de 90%, o un total de 0.9% con un rendimiento promedio de 80%. En contraste para una síntesis convergente que combina dos intermedios cada uno preparado por síntesis de 10 etapas (es decir, un total de 21 etapas), el rendimiento global será del 56% con un rendimiento promedio del 95%, o un 31% global con un rendimiento promedio del 90%, o un 9% global con un promedio del 80% rendimiento. En efecto, la síntesis convergente es de sólo 11 pasos. Las dos síntesis antes mencionadas son un caso concreto. El rendimiento promedio por etapa, 84-87%, en la segunda síntesis fue casi tan alto como el rendimiento promedio de 89% por paso en la primera síntesis. El rendimiento general 15 veces menor para la segunda síntesis es casi en su totalidad consecuencia de su mayor longitud, 29-35 pasos versus 21 pasos.
Construcción de cadena lateral estereotipada
Se deben cumplir dos retos estratégicos para completar una síntesis total de espatol (196). Primero, se presumió que el diepóxido vecinal alílico único en 196 era altamente electrófilo debido a que la apertura del anillo del epóxido por cloruro, un nucleófilo débil, acompaña a la esterificación tras el tratamiento de 196 con cloruro de p-bromobenzoilo y piridina. Segundo, los tres estereocentros de la cadena lateral flexible deben ensamblarse con las configuraciones correctas en relación con las del núcleo tricíclico rígido.
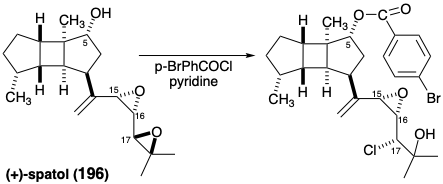
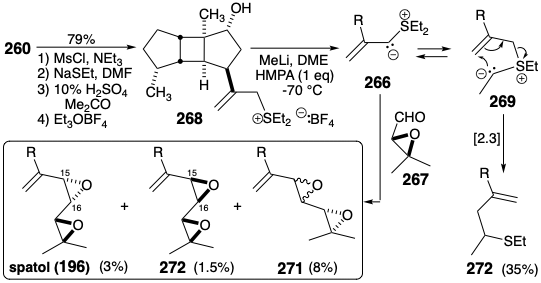
El grupo éster malónico en los intermedios (-) - 241 y 245 podría proporcionar un precursor alílico de tres carbonos de la cadena lateral de spatol. Koga convirtió 245 en alcohol alílico 260 mediante una reducción Marshall modificada.
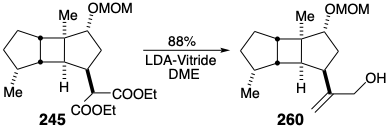
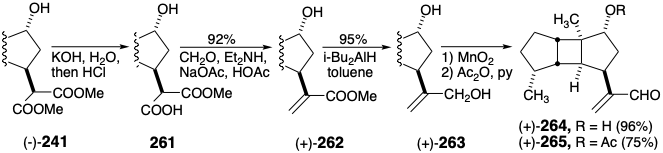
Sin embargo, el intento de conversión en un solo paso de (-) - 241 en el alcohol alílico 263 por la reducción Marshall, es decir, la reducción LAH del enolato de sodio, fracasó completamente. Por lo tanto, esta transformación se logró por monosaponificación a 261 y condensación aldólica descarboxilativa con formaldehído para proporcionar éster acrílico (+) - 262. La reducción de hidruro luego entregó alcohol alílico (+) - 263 que se oxidó selectivamente con\(\ce{MnO2}\) el aldehído (+) - 264. Para correlacionar este intermedio sintético con el producto natural, se acetiló (+) - 264. El acetato totalmente sintético mostró [α] D 22 +25.1° que se compara bien con el acetato de origen natural que mostró [α] D 22 +26.5°. 1
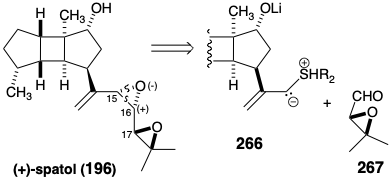
Una estrategia asimétrica absoluta
La desconexión tanto de un oxígeno nucleofílico como de un carbono electrófilo del carbono 15 de espatol sugiere un precursor 266 en el que el grupo funcional sulfonio proporciona la reactividad bifílica requerida en el carbono 15. 19 Las configuraciones relativas correctas para los estereocentros en el núcleo tricíclico y en la posición 17 se aseguran en una síntesis asimétrica absoluta mediante el uso de bloques 266 y 267 con las configuraciones absolutas correctas. Aunque muy breve, esta estrategia convergente no proporciona ningún control sobre las configuraciones en las posiciones 15 y 16.
El iluro 266 se preparó a partir del alcohol aliico 260 a través de la sal sulfonio 268. La reacción del iluro 266 con el aldehído 267 produjo spatol en solo 3% de rendimiento junto con el cis diepóxido isomérico alílico 270 (1.5%) y una mezcla de trans diepóxidos 271 (8%). Además, 266 existe en equilibrio con un iluro alternativo 269 que sufrió [2.3] reordenamiento sigmatropico produciendo el sulfuro homoalílico 272 (35%) como el producto principal de la reacción.
Una estrategia de reordenamiento estereoespecífico de epoxidiol
El último paso en la reacción del iluro 266 con el aldehído 264 implica la alquilación vecinal de un alcóxido durante la ciclación de 273. La reacción del aldehído 277 con iluro 276 es una estrategia relacionada. El aldehído epoxi 277 podría estar disponible a partir del aldehído (+) -264 por alquinilación de Corey-Fuchs para dar 278, homologación con formaldehído, reducción Lindlar del alcohol propargílico resultante, epoxidación asimétrica del alcohol alílico derivado y oxidación de Swern del resultante alcohol epoxídico. Alternativamente, 278 podría ser homologado a 275. Luego, después de la reducción asimétrica de esta propargilcetona, reducción de Lindlar y epoxidación catalizada por VO (acac) 2, la heterociclización de la 274 resultante podría entregar (+) -spatol. Sin embargo, estas estrategias son demasiado largas, y los cierres de anillos de intermedios como 274 pueden descarrilarse considerando el potencial, entre otras cosas, de eliminación de E-1 y transepoxidación. Así, el ataque intramolecular del alcóxido en 280 en la posición 2° 16 para dar 281 más bien que en la posición 3° 18 para dar espatol podría incluso ser favorecido. Tales reacciones de transepoxidación (reordenamientos de Payne) son bien conocidas. Sin embargo, el reordenamiento a 281 es reversible mientras que la heterociclización de 280 sería irreversible. Sin embargo, la eliminación de E1 para dar 279 parecía una preocupación razonable.
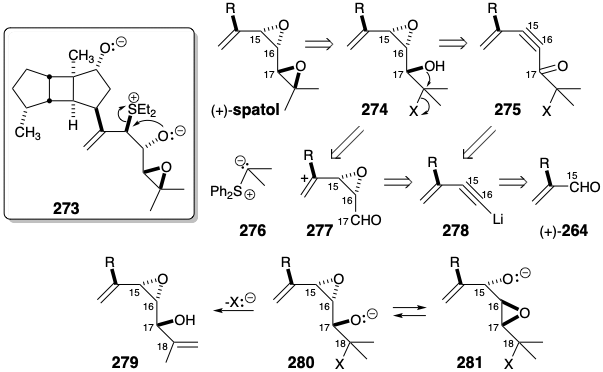
Una estrategia alternativa es posible que involucre un alcóxido epoxi similar al 281 pero con las posiciones del nucleófugo y el alcóxido intercambiadas. Así, el reordenamiento de Payne debería producir 283 pero la estereoquímica trans del epóxido en 282 prácticamente debería impedir el ataque directo del alcóxido en la posición 15 para producir un tetrahidrofurano. El electrófilo alílico en la posición 15 en 283 debería ser particularmente efectivo en la alquilación del alcóxido vecino produciendo un diepóxido alílico 284.
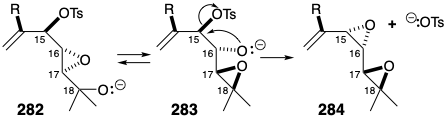
Además, una estrategia eficiente para ensamblar un precursor de epoxidiol 285 para 282 parecía factible. Así, 285 deberían estar disponibles por epoxidación regioselectiva de 286 la cual, a su vez, podría prepararse por la unión de un electrófilo C 15 (+) - 264 con un nucleófilo C 5 287.
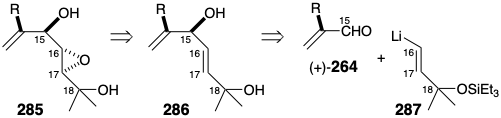
En estudios modelo, se buscó un método para producir la matriz de diepóxido alílico de la cadena lateral de spatol a partir de derivados apropiadamente activados del epoxidiol 285. Los resultados iniciales fueron decepcionantes. Así, la activación de eritro-288 como mesilato, eritro-289, seguido de tratamiento con sólido\(\ce{K2CO3}\) en isopropanol hirviendo suministró treo-290 por desplazamiento intermolecular de S N 2 en lugar del diepóxido vecinal deseado por Reordenamiento de Payne seguido de heterociclización. Dado que el grupo hidroxilo terciario en eritro-289 parecía no ser suficientemente nucleófilo para desplazar al grupo saliente epoxi, se buscaron condiciones que generaran un alcóxido a partir del hidroxilo terciario. El tratamiento con t-BuOK en t-BuOH promovió un reordenamiento limpio, estereoespecífico y heterociclización para suministrar el diepóxido trans, eritro-292. Una ruta del eritro-288 a un derivado activado del alcohol treo epoxi requiere activación con inversión concomitante de configuración. Esto se logró mediante la modificación Still de la reacción de Mitsunobu. Así, el tratamiento de eritro-288 con\(\ce{Zn(OTs)2}\) azodicarboxilato de dietilo y trifenilfosfina, dio tosilato treo-291 que, tras el tratamiento con t-BuOK en DMF, proporcionó el diepóxido alílico cis, eritro-292 con 82% de rendimiento.
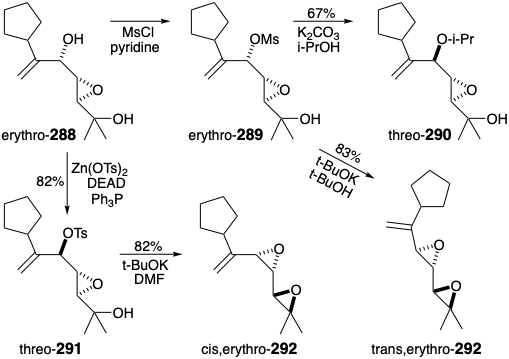
La reacción de sustitución del mesilato eritro-289 con isopropanol para dar treo-290 sugiere que una sustitución similar con hidróxido de tetrabutilamonio podría proporcionar una ruta al alcohol invertido. En cambio, sin embargo, se obtuvo un alto rendimiento de diepóxido trans, eritro-292. La inesperada estabilidad de este diepóxido alílico frente al hidróxido es especialmente interesante en vista de la reacción de sustitución de escisión de epóxido de spatol con el anión cloruro menos nucleófilo que da una clorohidrina (véase supra). Al parecer, esta última reacción es una apertura de epóxido catalizada por ácido inducida por clorhidrato de piridinio, subproducto de la acilación con cloruro de p-bromobenzoilo en presencia de piridina.
En un primer intento de implementar el plan, la adición de vinillitio 287 al aldehído (+) - 264 proporcionó una mezcla 1:6 de trioles 295 y 296 respectivamente después de la desililación de los éteres monosililo intermedios 293 y 294. Para controlar la regioselectividad de la epoxidación, el triol 296 se disililo selectivamente. La epoxidación catalizada con vanadio de 297 fue dirigida al enlace 15,16-C=C por el resto del hidroxilo alílico. Dado que el producto epóxido principal fue el eritro derivado 298e, la activación selectiva del 15-hidroxilo menos impedido en el correspondiente triol 298e se realizó con inversión de configuración. Sin embargo, el tratamiento con base produjo un diepóxido alílico 300 con un núcleo cis, anti, cis-triciclo [5.3,0,0 2,5] decano. Así, el reordenamiento Wagner-Meerwein del núcleo cis, anti, cis-triciclo [5,3,0,0 2,6] decano de 298e para dar 300, aparentemente debido a una activación no intencionada del 5-hidroxilo que acompañó a la activación deseada del hidroxilo en el 15 posiciones.
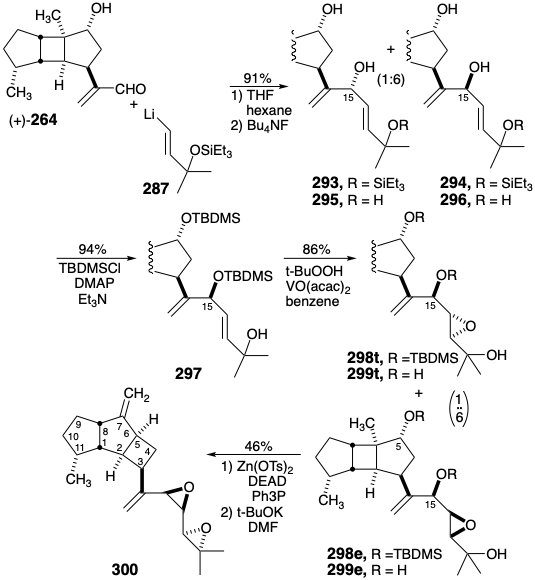
Estos resultados sugirieron que los derivados de los epoxi trioles 299e y 299t en los que el hidroxilo en la posición 5 está enmascarado fueron necesarios para la generación de la cadena lateral de espatol sin el reordenamiento acompañante del núcleo tricíclico. La labilidad de la matriz de diepóxidos alílicos en spatol (196) bajo condiciones ácidas limitó la elección de derivados a aquellos con grupos enmascarantes que serían removibles en condiciones neutras o básicas de reacción. El requisito adicional de estabilidad hacia un reactivo de vinillitio y la presencia de insaturación en la diana sintética recomendaron derivados de éter de p-metoxibencilo (MBn). El grupo de enmascaramiento MBn es removible en condiciones suaves por escisión oxidativa con DDQ. Por lo tanto, los derivados de MBn eritro - 308b y treo - 308b de 298e y 298t se prepararon a partir de diol (+) - 263.
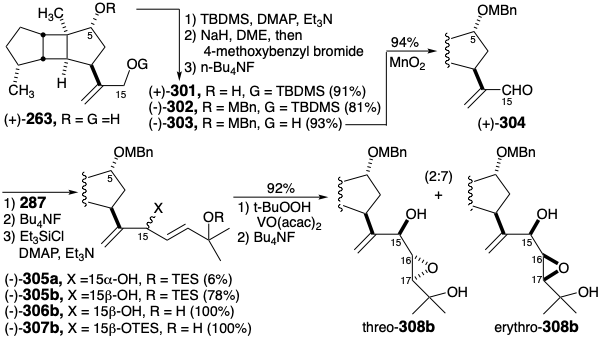
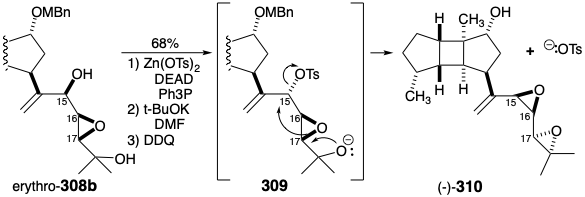
La asignación de una configuración absoluta S en la posición 15 al epímero mayor (-) - 305b se basó en la correlación con el espatol natural (+) (vide infra). La epoxidación del silil éter derivado (-) - 307 proporcionó una mezcla 2:7 de treo y eritro epóxidos 308b. El isómero principal, eritro-308b, se convirtió en un cis, eritro diepóxido (-) - 310 por conversión a un treo tosilato con configuración invertida en la posición 15 seguido de transposición de Payne inducida por bases, heterociclización y finalmente por desprotección del 5-hidroxilo. Que (-) - 310 no era espatol (196) fue evidente por su actividad óptica, [α] D = -10.0° en contraste con [α] D = +45.6° reportado para el producto natural. Pequeñas diferencias de desplazamiento químico, por ejemplo resonancias de RMN de vinilo 1H a δ5.14 y 5.09, confirmaron que (-) - 310 es epimérico en las posiciones 15, 16 y 17 con (+) -spatol que exhibe resonancias vinílicas a δ5.13 y 5.02.
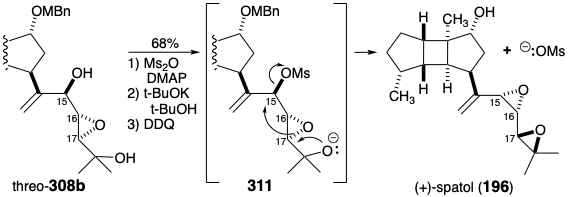
El isómero menor, treo-308b, se convirtió en (+) -spatol (196) por monomesilación seguida de transposición de Payne inducida por bases, heterociclización y desprotección del 5-hidroxilo. Cada resonancia en el espectro de RMN 1H de (+) -spatol sintético coincidió dentro de 0.01 ppm con un espectro de una muestra auténtica de espatol natural.
Una estrategia estereoconvergente asimétrica absoluta
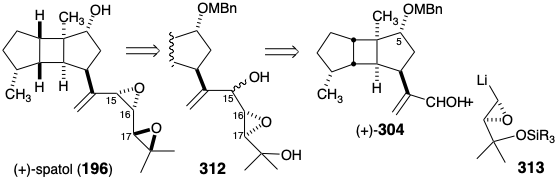
Ambas estrategias antes mencionadas para el diepóxido alílico de espatol presentan un estereocontrol inadecuado. Así, mientras que la configuración absoluta correcta en la posición 17 en 273 se asegura usando el enantiómero correcto de 267, la generación de los estereocentros en las posiciones 15 y 16 en 273 no es selectiva. De manera similar, aunque cualquiera de los epímeros en la posición 15 en 308 puede proporcionar un derivado activado con la configuración correcta, es decir, por activación con retención o inversión de configuración, la generación de los estereocentros en las posiciones 16 y 17 en 308 no es favorablemente selectiva.
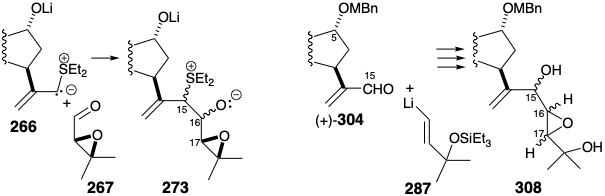
Dado que cualquiera de los epímeros del epoxidiol 312 se puede convertir estereoespecíficamente en spatol, la síntesis es estereoconvergente y el estereocontrol en la posición 15 es innecesario para una síntesis total eficiente. La estereoquímica correcta en las posiciones 16 y 17 podría ser asegurada por una estrategia asimétrica absoluta que combina el aldehído homoquiral C 15 (+) - 304 y un nucleófilo homoquiral C 5 α-epoxi 313.
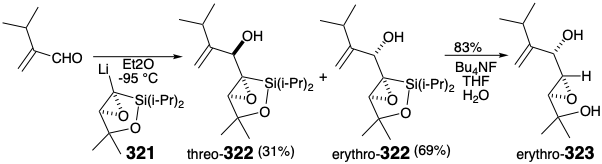
Debido a que los epóxidos de α-sililo son fácilmente hidrodesililados por fluoruro húmedo con retención completa de la configuración, un posible equivalente sintético del sintón 313 es el α-litioepóxido 314 estabilizado con sililo. No obstante, un grave defecto podría sabotear esta estrategia. Así, aunque los α-litioepóxidos son generalmente configuracionalmente estables, el α-litioepóxido 316, un análogo cercano de 314, exhibe una inestabilidad configuracional inusual reordenándose completamente a 317 debido, sin duda, a la tensión estérica que se alivia en trans-cis isomerización. Una isomerización similar de 314 a 315 descarrilaría la síntesis. Este escollo fue eludido por un puente temporal entre el sustituyente C-sililo y el oxígeno vecino en 321. Así, la O-sililación intramolecular impide la isomerización del carbanión 321 obtenido por metalación de 320. El racémico 321 se preparó mediante epoxidación de un vinilsilano 319 que se generó por una nueva hidrogenación-deshidrogenación-heterociclización de 318.
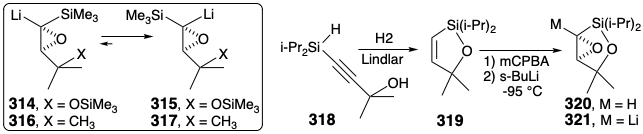
En un estudio modelo, la reacción de α-litioepóxido 321 con 2- (i-propil) acroleína liberó una mezcla epimérica de aductos 322 favoreciendo el eritro diastereómero por 7:3. La desililación de eritro- 322 dio epoxidiol eritro- 323. Así, el intermedio racémico 321 proporciona una síntesis en dos etapas de precursores de epoxidiol del diepóxido alílico de spatol. Sin embargo, solo la conjunción del aldehído (+) - 304 con el enantiómero correcto de 321 proporcionará las configuraciones absolutas correctas en las posiciones 16 y 17 en 312 que se requieren para lograr una construcción eficiente de espatol natural (196). Aún se debe encontrar una ruta hacia el epóxido 321 ópticamente puro.