10.1: Reacciones Quirales Basadas en Prolina
- Page ID
- 73158

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)El organocatálisis enantioselectivo ha surgido como un potente método sintético complementario a las reacciones catalizadas por metales y enzimas. La baja toxicidad asociada con la organocatálisis y la simplicidad operativa lo convierten en un método atractivo para sintetizar estructuras complejas. Entre los organocatalizadores, pequeñas moléculas como prolina quiral, tiourea quiral, TADDOL quiral y alcaloides quirales tienen una reactividad especial en la síntesis asimétrica.
La prolina quiral se denomina como los organocatalizadores bifuncionales más simples (Esquema\(\PageIndex{1}\)). Este aminoácido se denomina como “enzima más simple” debido a su capacidad para catalizar reacciones con alta estereoselectividad.
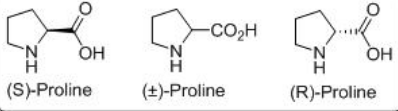
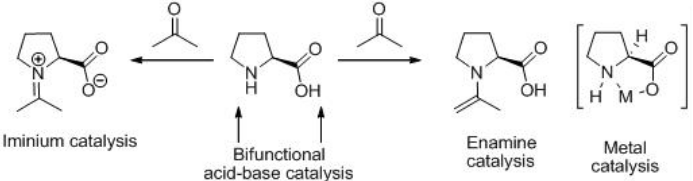
La L-prolina es una molécula pequeña, no tóxica, económica, fácilmente disponible en ambas formas enantioméricas que tiene sitios ácido-base bifuncionales (Esquema\(\PageIndex{2}\)). La reacción puede proceder a través de catálisis de iminio, catálisis de enamina o catálisis bifuncional ácido-base.
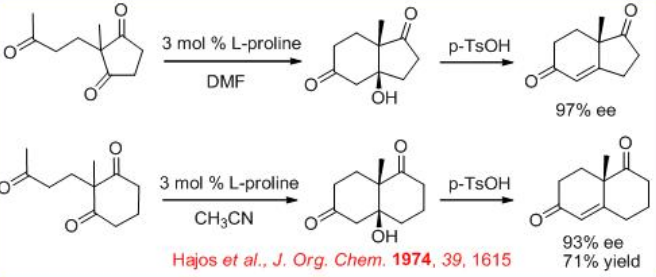
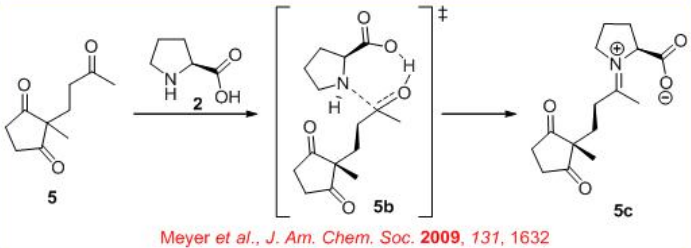
A principios de 1970 apareció la primera ciclación aldólica catalizada por L-prolina (Esquema\(\PageIndex{3}\)). Después de casi 25 años, se ha ilustrado el estado de transición esperado para la reacción (Esquema\(\PageIndex{4}\)).
Reacción de Aldol Intermolecular
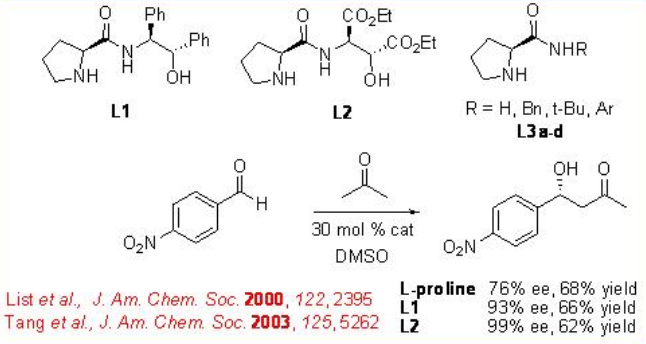
La reacción aldólica enantioselectiva es uno de los métodos más potentes para la construcción de poliol quiral. La primera reacción aldólica enantioselectiva directa intermolecular catalizada por L-prolina apareció empleando acetona y 4-nitrobenzaldehído como sustratos (Esquema\(\PageIndex{5}\)). Este resultado despertó un gran interés de varios grupos en investigar más a fondo las reacciones aldólicas asimétricas directas catalizadas por prolina. Posteriormente, se han desarrollado catalizadores derivados de prolina quiral modificados L1-3 para potenciar la selectividad de la reacción.
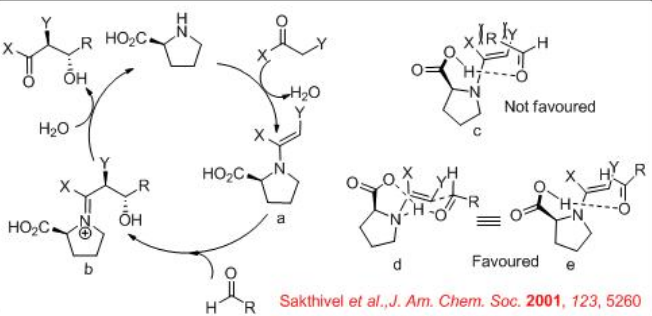
Para el mecanismo, la reacción de pirrolidina con el donador de carbonilo puede dar enamina a que podría proceder a la reacción con la cara re de los aldehídos para dar el ion iminio b (Esquema\(\PageIndex{6}\)). Este último puede someterse a hidrólisis para proporcionar β-hidroxicetona quiral. El estado de transición propuesto ilustra que el ataque de enamina ocurre en la cara re del aldehído d y e. Esta selectividad facial de ataque por la enamina viene dictada por minimizar las interacciones estéricas entre el sustituyente aldehído y el sustituyente enamina. El ataque de la enamina en la cara si del aldehído conduce al estado de transición desfavorable c.
Reacción de Mannich
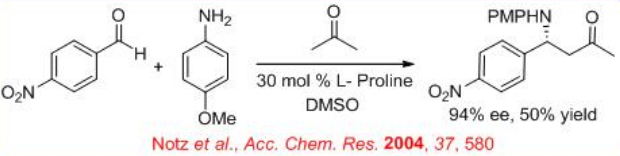
Paralelamente a la reacción aldólica, se ha explorado la reacción enantioselectiva de Mannich de aldehído, acetona y p-anisidina como sustratos con 50% de rendimiento y 94% ee (Esquema\(\PageIndex{7}\)).
El mecanismo es análogo al de las reacciones aldólico (Esquema\(\PageIndex{8}\)). La reacción de prolina con aldehído o cetona puede dar enamina que podría experimentar reacción con la imina para formar nuevos estereocentros como producto de iminio. Este último en la hidrólisis puede dar el producto Mannich objetivo. La reacción de (E) -aldimina con la enamina en su cara si puede dar el producto syn. Debido a que la cara re está bloqueada por interacciones estéricas entre el anillo aromático del grupo p-metoxifenilo y el anillo de prolina.
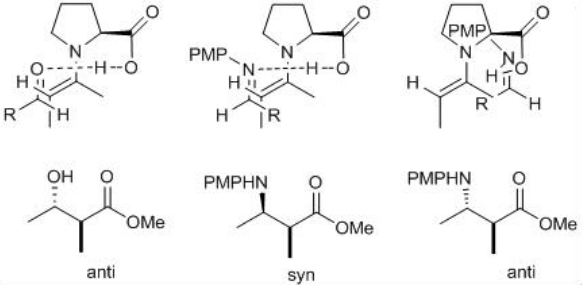
Las reacciones de Mannich catalizadas por prolina de α-imino etil glioxilato protegido con N-PMP con una variedad de cetonas proporcionan α-aminoácidos funcionalizados (Esquema 9). Estas reacciones pueden generar dos centros estereogénicos adyacentes simultáneamente tras la formación de enlaces C-C con estereocontrol sin completo y pueden realizarse en una escala de gramos con simplicidad operativa.
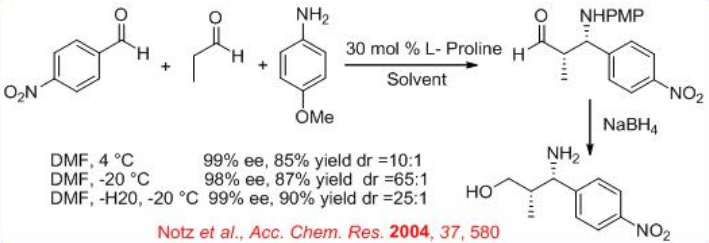
La reacción catalizada por prolina del glioxilato de α-imino etilo protegido con N-PMP con aldehídos alifáticos proporciona un método general para la síntesis de derivados β-amino y α-aminoácidos (Esquema\(\PageIndex{10}\)). La diastereoselectividad depende de la voluminosidad de los sustituyentes del donante de aldehído. En la mayoría de los casos se puede lograr una alta estereoselectividad sin.
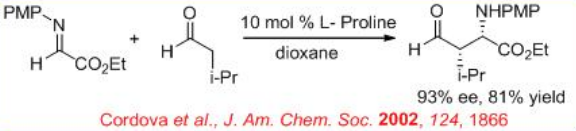
La síntesis de derivados de aminoácidos cuaternarios quirales se puede lograr usando catálisis basada en prolina (Esquema\(\PageIndex{11}\)). El nitrógeno se une a la α-aril amina con el fin de aumentar la reactividad a través de la cepa de anillo y los productos se obtienen con alta enantioselectividad.
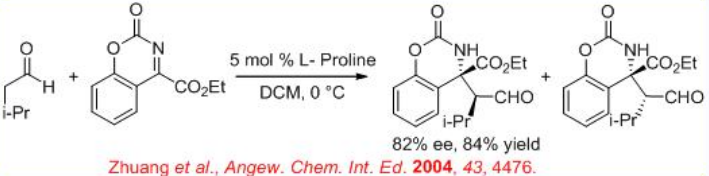
La reacción de tipo Mannich catalizada por (S) -prolina de aldehídos con glioxilato de α-imino etilo proporciona productos sin, mientras que la reacción que utiliza ácido (3 R, 5 R) -5-metil-3-pirrolidinacarboxílico da producto antiselectivo (Esquema\(\PageIndex{12}\)).
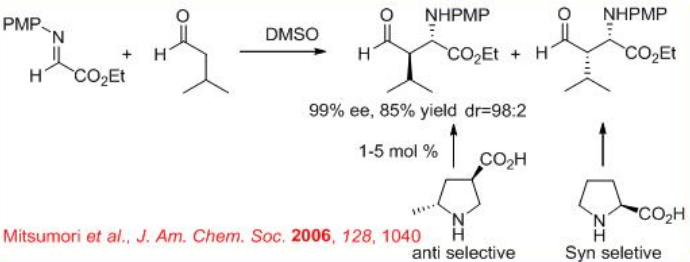
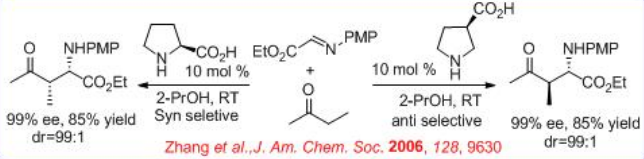
Además, el ácido (R) -3-pirrolidinacarboxílico cataliza las reacciones de tipo Mannich de cetonas con glioxilato de α-imino etilo para dar antiproductos, mientras que las reacciones basadas en (S) - prolina dan productos syn (Esquema\(\PageIndex{13}\)). Así, la posición del grupo ácido carboxílico en el anillo de pirrolidina dirige la estereoselección de la reacción catalizada proporcionando productos syn - o anti-Mannich.
Michael Reacción
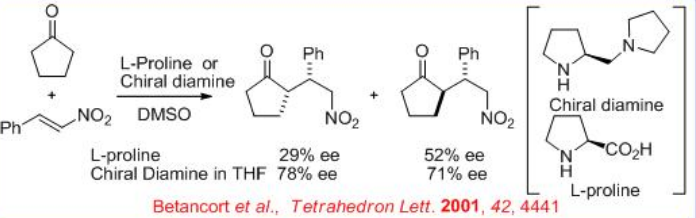
En 2001 apareció el primer ejemplo de una reacción asimétrica directa de Michael empleando un donante activado por enamina. La reacción catalizada por prolina de acetona y ciclopentanona con benzalmalonato y nitroestireno proporciona el producto Michael con bajo exceso enantiomérico. Sin embargo, el uso de diamina quiral mejora significativamente el ee con nitroestireno y malonatos de alquilideno como aceptores y donantes de cetonas (Esquema\(\PageIndex{14}\)).
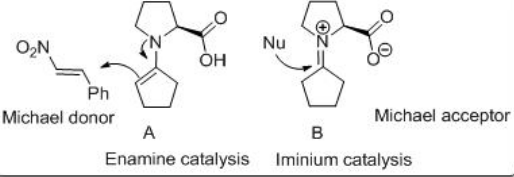
El posible resultado estereoquímico se ha contabilizado asumiendo los estados de transición acíclicos A y B. Estas reacciones de Michael constituyeron las primeras reacciones asimétricas catalíticas directas de cualquier tipo con donantes de aldehído y fomentaron el desarrollo de reacciones basadas en aldehído con una gama de electrófilos (Esquema\(\PageIndex{15}\)).
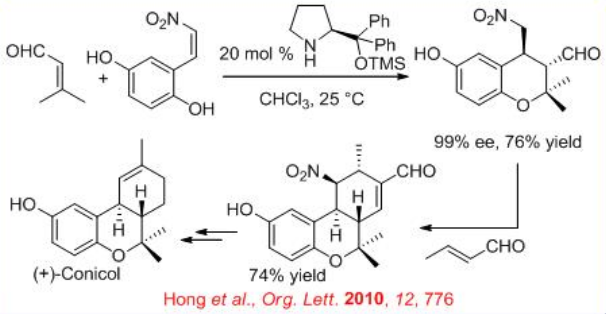
Se puede prever el modo de activación de iminio-enamina para explicar la reacción dominó Oxa-Michael—Michael que ocurre entre 3-metilbut-2-enal y (E) -2- (2-nitrovinil) -benceno-1,4-diol tras la catálisis con difenil prolinol silil éter quiral, que proporcionan el correspondiente enantiopuro Oxa-Michael— Cicloaducto de Michael en 76% de rendimiento y 99% ee (Esquema\(\PageIndex{16}\)). Este último puede estar implicado adicionalmente en una secuencia de Michael-Aldol a través de la reacción con crotonaldehído para proporcionar el correspondiente hexahidro-6H-benzo-cromeno con 74% de rendimiento. Estas dos reacciones dominó han constituido los pasos clave de la primera síntesis total asimétrica del producto natural biológicamente activo (+) -conicol.