10.3: Catálisis a base de tiourea
- Page ID
- 73149

Síntesis de Strecker
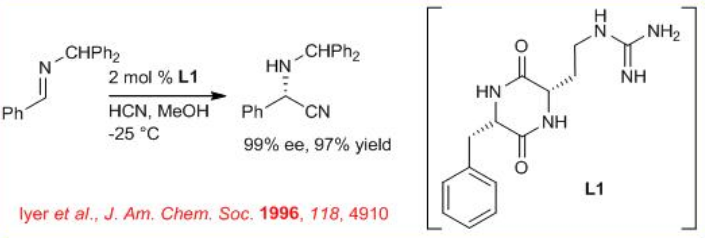
En 1996 apareció la primera síntesis organocatalítica asimétrica de Strecker empleando L1 como catalizador (Esquema\(\PageIndex{1}\)). La reacción implica la adición de HCN a iminas en presencia de derivado dicetopiperazina con hasta > 99% ee.
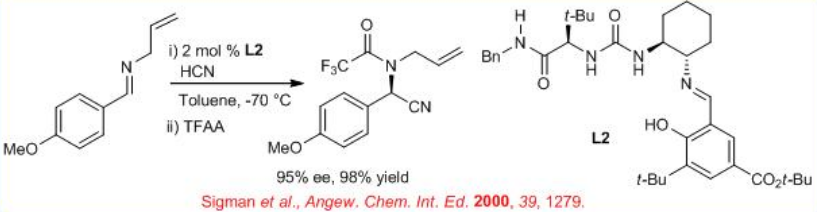
Posteriormente, se ha utilizado el derivado de tiourea quiral L2 para esta reacción para proporcionar las cianohidrinas con 98% ee (Esquema\(\PageIndex{2}\)).
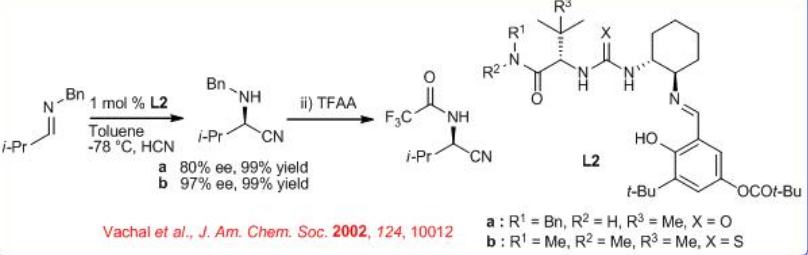
Se han realizado mejoras adicionales en esta reacción empleando el derivado de tiourea L3 (Esquema\(\PageIndex{3}\)). El sitio activo del catalizador, el estereoisómero relevante del sustrato de imina y la estructura en solución del complejo imina-catalizador se dilucidan mediante cinética, actividad estructural y experimentos de RMN. Se identifica una inusual interacción puente entre la imina y los hidrógenos de urea del catalizador.
Reacción de Mannich
En paralelo a la reacción de Strecker, se estudia la reacción de Mannich de una amplia variedad de N-Boc aril iminas en presencia del derivado de tiourea L3 con alta enantioselectividad (Esquema\(\PageIndex{4}\)). El catalizador L3 es altamente efectivo para la adición asimétrica de derivados de silil ceteno acetal a aldiminas. Desde un punto de vista estérico y electrónico, los sustratos de N-Boc imina utilizados en esta reacción son fundamentalmente diferentes de los derivados N-alquílicos empleados en la reacción de Strecker.
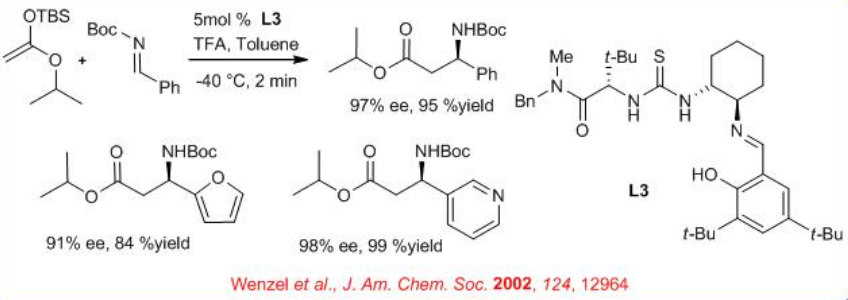
El derivado bifuncional de tiourea L4 puede catalizar la reacción de Michael de malonatos con diversas nitroolefinas en alta enantioselectividad (Esquema\(\PageIndex{5}\)). El catalizador activa nucleófilo por catálisis básica general y electrófilo por unión H al grupo nitro. Esta metodología se ha aplicado para adiciones enantioselectivas de cetoéster sustituido y adiciones dobles de Michael de cetoésteres α, β -insaturados.
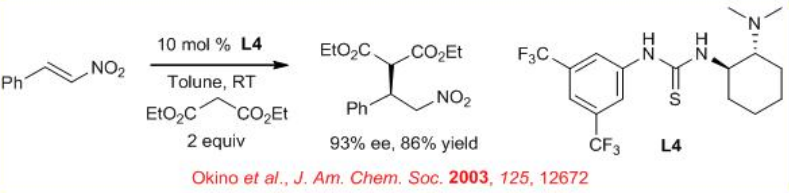
La amina primaria quiral - tiourea L5 es efectiva para la adición directa conjugada de cetonas a nitroalquenos (Esquema\(\PageIndex{6}\)). La anti diastereoselectividad observada sugiere la participación de un intermedio (Z) -enamina que es complementario a la diastereoselectividad obtenida en reacciones análogas que involucran (E) -enaminas generadas a partir de catalizadores de amina secundaria.
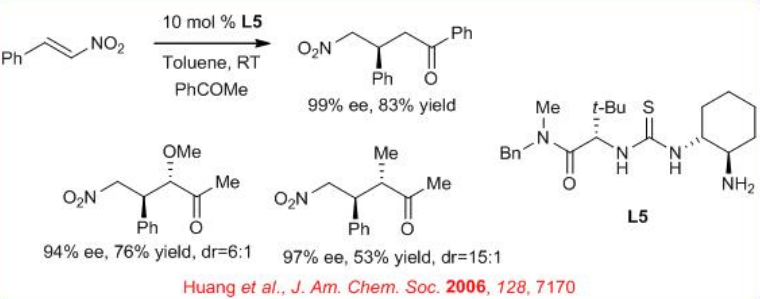
Asimismo, se ha demostrado la adición de un rango de nitroalcanos a N-Boc iminas aromáticas utilizando el derivado de tiourea L6 con mayormente anti diastereoselectividad (Esquema\(\PageIndex{7}\)).
Se ha revelado que el catalizador de tiourea L7 que lleva grupos 3,5-bis (trifluorometil) benceno y dimetilamino es eficiente para la reacción asimétrica de Michael de compuestos 1,3-dicarbonilo a nitroolefinas (Esquema\(\PageIndex{8}\)). Esta metodología se ha aplicado para la síntesis total de (R) - (−) -baclofeno. La reacción de 4-cloronitroestireno y compuesto 1,3-dicarbonilo genera centro de carbono cuaternario con 94% ee. La reducción del gruop nitro a amina y la posterior ciclación, esterificación y apertura del anillo proporciona (R) - (−) -baclofeno en 38% de rendimiento.
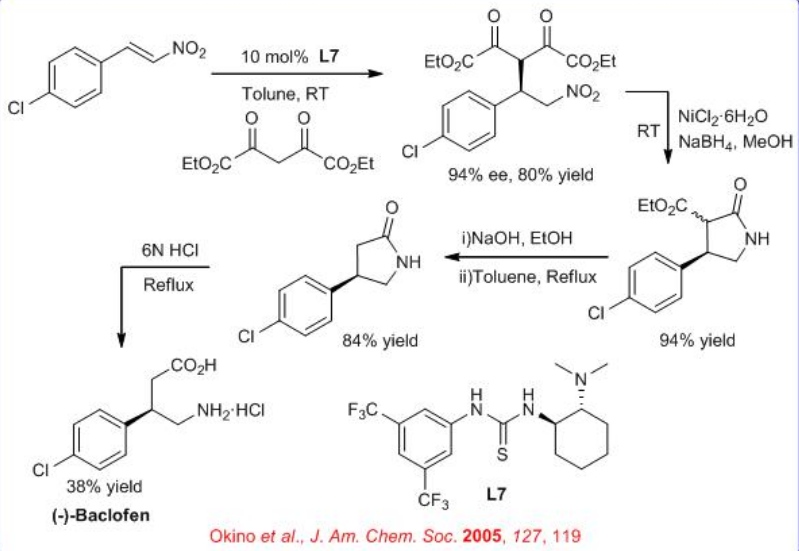
El mecanismo de la adición enantioselectiva anterior de Michael de acetil acetona a una nitroolefina catalizada por un organocatalizador quiral bifuncional basado en tiourea ha sido investigado usando cálculos de la teoría funcional de la densidad y los resultados sugieren que ambos sustratos se coordinan preferentemente vía bidentado enlaces de hidrógeno (enlace H) (Esquema\(\PageIndex{9}\)). Se encuentra que la desprotonación de la forma enol de la acetilacetona por la amina del catalizador se produce fácilmente, lo que lleva a un par iónico caracterizado por múltiples enlaces H que involucran también a la unidad de tiourea. Se han explorado dos vías de reacción distintas hacia la formación del producto Michael que difieren en el modo de activación electrófila. Se ha demostrado que ambos canales de reacción son consistentes con la noción de organocatálisis no covalente, ya que los estados de transición que conducen al aducto de Michael se estabilizan mediante extensas redes unidas por H.
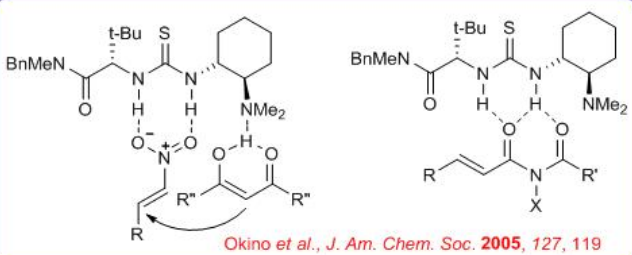
Se ha desarrollado una adición de Michael asimétrica catalizada por tiourea de compuestos de metileno activados a imidas α, β- insaturadas (Esquema\(\PageIndex{10}\)). La N-alquenoil-2-metoxibenzamida es el mejor sustrato entre los derivados de benzamida correspondientes que portan diferentes sustituyentes en el anillo aromático y reaccionan con varios compuestos de metileno activados como malononitrilo, metil α-cianoacetato y nitrometano con hasta 93% ee. La reactividad puede atribuirse a la interacción intramolecular de enlace H entre el N-H de la imida y el grupo metoxi del grupo benzamida.
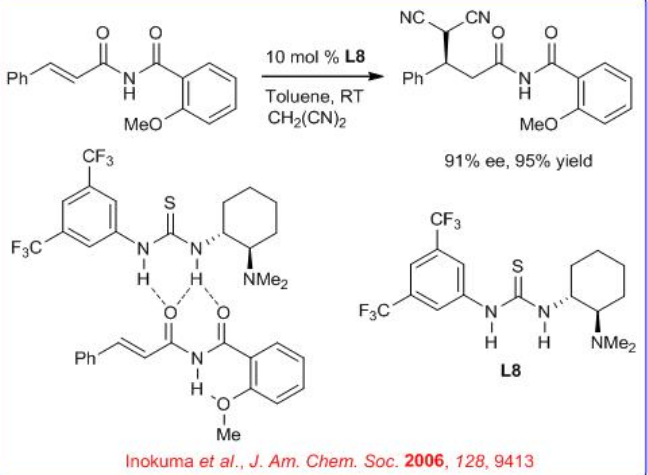
El catalizador de tiourea L9 ha sido explorado para la activación de quinolina con ácidos organoborónicos para facilitar el estereocontrol en la transformación de Petasis incluso a bajas temperaturas (Esquema\(\PageIndex{11}\)). La quinolina se activa mediante la formación de N-COBz con PhCOCl y se puede lograr un alto grado de control estéreo usando una combinación de H 2 O y NaHCO 3 como aditivos.
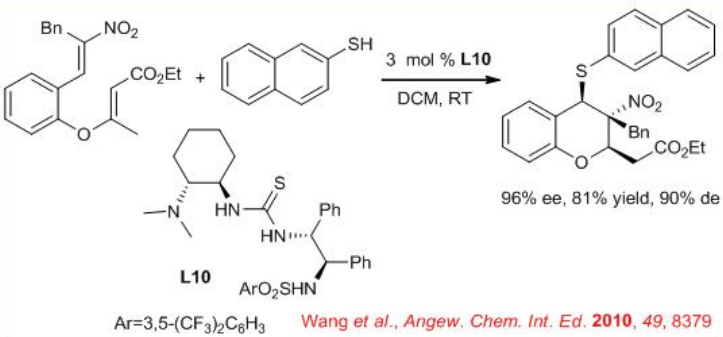
La reacción dominó Thia-Michael-Michael de tioles con enoatos de nitroolefina proporciona derivados de cromano polifuncionalizados de manera altamente estereoselectiva en presencia de tiourea L10. Se pueden generar tres centros estereogénicos consecutivos incluyendo un estereocentro cuaternario con alta enantioselectividad (Esquema\(\PageIndex{12}\)). El catalizador L10 activa los enoatos de nitroolefina a través de la activación de enlaces H, y su fracción amino terciaria activa los tioles nucleofílicos, formando un intermedio que experimenta la adición intermolecular de tia-Michael.
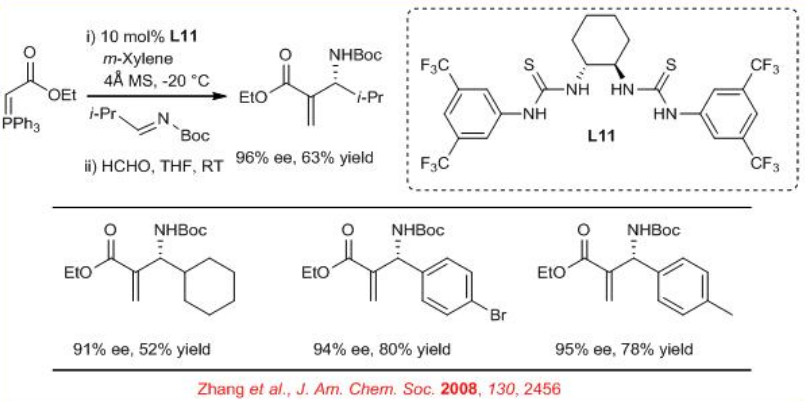
La síntesis de ésteres quirales N-Boc- β-amino- α -metilen carboxílicos se puede realizar por reacción de iluros de fósforo estabilizados y aldiminas protegidas con Boc en presencia de bistiourea L11 fácilmente disponible (Esquema\(\PageIndex{13}\)). La reacción posterior con formaldehído proporciona un fácil acceso a ésteres quirales de N - Boc- β-amino- α -metileno carboxílicos. Se ha encontrado que el catalizador es reciclable.
Reacciones de hidrofosfonilación
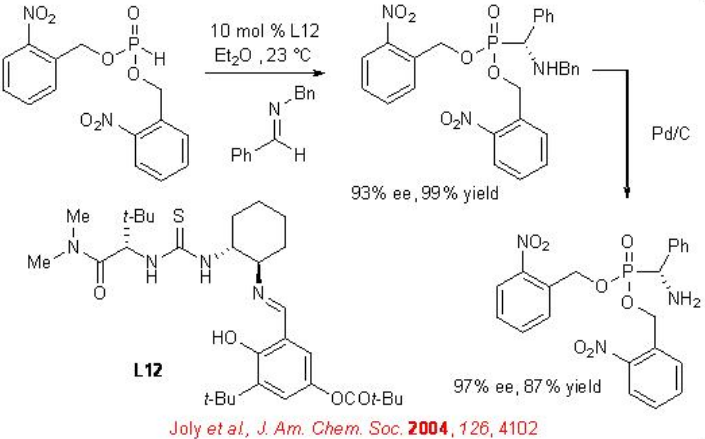
El catalizador de tiourea quiral L12 se ha utilizado para la hidrofosfonilación altamente enantioselectiva de una amplia gama de N-bencil iminas (Esquema\(\PageIndex{14}\)). Los productos hidrofosfonilados pueden desprotegerse fácilmente por hidrogenólisis usando Pd/C para proporcionar ácidos α-amino fosfónicos quirales con alta enantioselectividad. Esta metodología proporciona un acceso general y conveniente para la síntesis de α-amino fosfonatos ópticamente activos.