
8.8: Reacciones de Sustitución Nucleofílica Biológica
- Page ID
- 72691

Las reacciones de sustitución nucleofílica que hemos visto hasta ahora han sido todas reacciones de laboratorio, más que bioquímicas. Ahora, finalmente, echemos un vistazo a algunos ejemplos de sustituciones nucleofílicas en un contexto biológico. Todos los principios que hemos aprendido hasta ahora todavía se aplican a estas reacciones bioquímicas, pero además hay que considerar los papeles de los catalizadores enzimáticos.
Esta es la primera vez que estaremos viendo mecanismos de reacción orgánica biológica 'reales'. No se deje intimidar por el tamaño y complejidad de las biomoléculas reaccionantes; son solo moléculas orgánicas, con los mismos patrones de unión y grupos funcionales con los que ya está familiarizado. Centrarse en las partes reaccionantes de la molécula: ¿qué es el nucleófilo? ¿El electrófilo? ¿El grupo que se va? En la mayoría de las reacciones orgánicas biológicas, la mayor parte de la biomolécula es simplemente 'ir a lo largo', y a menudo se puede abreviar con un 'grupo R' (sección 1.2) para simplificar la imagen.
Una\(S_N2\) reacción bioquímica
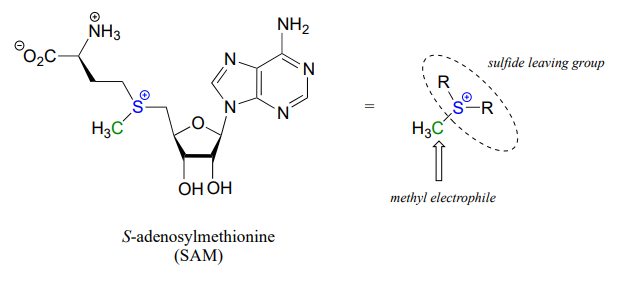
Una clase muy importante de reacciones de sustitución nucleofílica en bioquímica son las\(S_N2\) reacciones catalizadas por enzimas metiltransferasa dependientes de\(S\) -adenosil metionina (SAM). SAM es una coenzima (sección 6.3) que desempeña el papel de donante del grupo metilo: se puede pensar en SAM en este contexto como simplemente un electrófilo de metil carbono unido a un grupo saliente de sulfuro.
Hay muchas variaciones de reacciones de metilación dependientes de SAM en la naturaleza. En la introducción a este capítulo, se nos presentó una reacción que ocurre en el ADN bacteriano en la que un carbono metílico se transfiere de SAM a un átomo de nitrógeno sobre la adenina (este tipo de reacción a menudo se conoce como\(N\) -metilación).
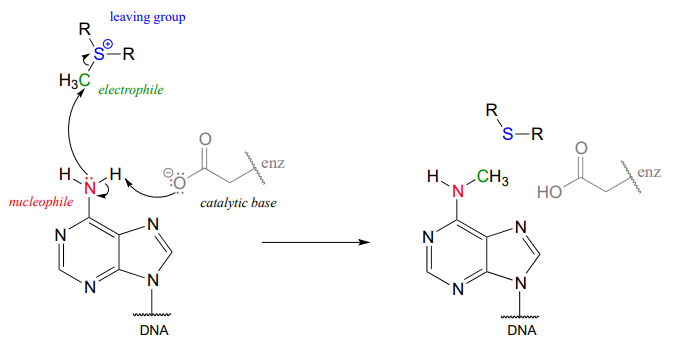
En la figura anterior, se muestra cómo un residuo de aspartato en el sitio activo de la enzima actúa como base catalítica: la transferencia de un protón desde el sustrato a la cadena lateral de aspartato comienza a potenciar la nucleofilia del nitrógeno amínico a medida que se acerca al carbono metílico electrófilo de SAM, y la formación del nuevo\(N-C\) vínculo y comienza la escisión del\(C-S\) vínculo. Estos cuatro eventos de reorganización de bonos probablemente se llevan a cabo de manera concertada. Un estado de transición probable se aproxima a continuación:
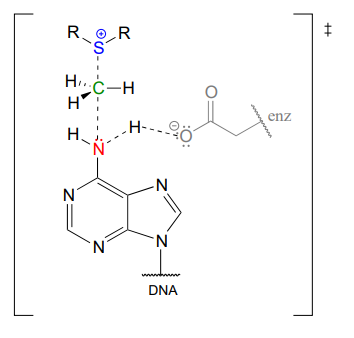
Por supuesto, existen muchas otras interacciones no covalentes entre los residuos enzimáticos del sitio activo y el sustrato (la base adenina) y el cofactor (SAM), pero en aras de la claridad estos no se muestran. Estas interacciones, muchas de las cuales son enlaces de hidrógeno, ayudan a posicionar la base de adenina y SAM en la orientación relativa correcta dentro del sitio activo para que tenga lugar el ataque nucleofílico. (Si tiene acceso a revistas de la American Chemical Society, un artículo sobre una enzima que cataliza una reacción de N-metilación similar contiene algunas cifras detalladas que muestran interacciones de enlace de hidrógeno y carga-dipolo entre el sitio activo de la enzima y los dos sustratos: ver Biochemistry 2003, 42, 8394, figura 4).
El electrófilo es un carbono metílico, por lo que hay poco obstáculo estérico para frenar el ataque nucleofílico. El carbono es electrófilo (pobre en electrones) porque está unido a un azufre cargado positivamente, que es un poderoso grupo aceptor de electrones. La carga positiva sobre el azufre también lo convierte en un excelente grupo saliente, ya que a medida que sale, se convierte en un sulfuro neutro y muy estable. Con todo, tenemos un buen nucleófilo (potenciado por la base catalítica), un electrófilo sin obstáculos y un excelente grupo saliente. Podemos predecir con confianza que esta reacción es\(S_N2\). Un\(S_N1\) mecanismo es extremadamente improbable: un catión metílico es muy inestable y por lo tanto no es un intermedio razonable para proponer.
Observe algo más sobre el mecanismo de metilación SAM ilustrado en la figura anterior. Es termolecular: hay tres actores actuando en concierto: la base catalítica, el nucleófilo y el electrófilo. Esto es posible porque los tres jugadores están unidos en una geometría muy específica en el sitio activo de la enzima. En una reacción que tiene lugar libre en solución, más que en un sitio activo, la probabilidad de que tres moléculas separadas colisionen todas a la vez, con la geometría justa para que se produzca una reacción, es muy, muy baja. Deberías notar en el futuro que cuando ilustramos el mecanismo de una reacción que tiene lugar libre en solución, solo veremos pasos bimoleculares: dos moléculas colisionando. Casi todas las reacciones bioquímicas que vemos en este libro serán catalizadas por enzimas -y los pasos termoleculares serán comunes- mientras que casi todas las reacciones de laboratorio que vemos se llevarán a cabo libres en solución, por lo que solo veremos pasos unimoleculares y bimoleculares. (Los químicos sintéticos suelen emplear catalizadores no biológicos que imitan sitios activos enzimáticos, pero estos ejemplos están mucho más allá del alcance de nuestra discusión).
Piense en el capítulo ácido-base: el\(pK_a\) de un éter protonado es aproximadamente cero, lo que indica que un éter es una base muy débil. Considerando las tendencias periódicas en acidez y basicidad, ¿qué se puede decir sobre la basicidad relativa de un sulfuro?
Otra reacción de metilación dependiente de SAM es catalizada por una enzima llamada catecol\(O\) - metiltransferasa. El sustrato aquí es la epinefrina, también conocida como adrenalina, y la reacción es parte de la vía por la cual la adrenalina se degrada en el cuerpo.
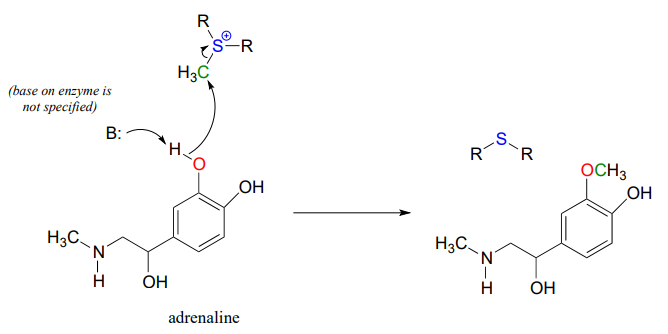
Observe que en este ejemplo, el nucleófilo atacante es un fenol oxígeno más que un nitrógeno (por eso la enzima se llama\(O\) -metiltransferasa). En muchos casos al dibujar mecanismos de reacción bioquímica, utilizamos las abreviaturas B: para una base catalítica y\(H-A\) para un ácido catalítico, para evitar que los dibujos se pongan demasiado 'ocupados' (también es posible que no se conozca la identidad del grupo ácido o básico).
La SAM está formada por una reacción de sustitución nucleofílica entre metionina y trifosfato de adenosina (ATP). Dibuja un mecanismo para esta reacción y explica por qué elegiste una\(S_N2\) vía\(S_N1\) o y.
Una\(S_N1\) reacción bioquímica
Como veremos en el capítulo 10,\(S_N1\) las reacciones catalizadas por enzimas juegan un papel crítico en el metabolismo de los carbohidratos y los nucleótidos de ADN/ARN. La siguiente reacción es parte de la biosíntesis de nucleótidos:
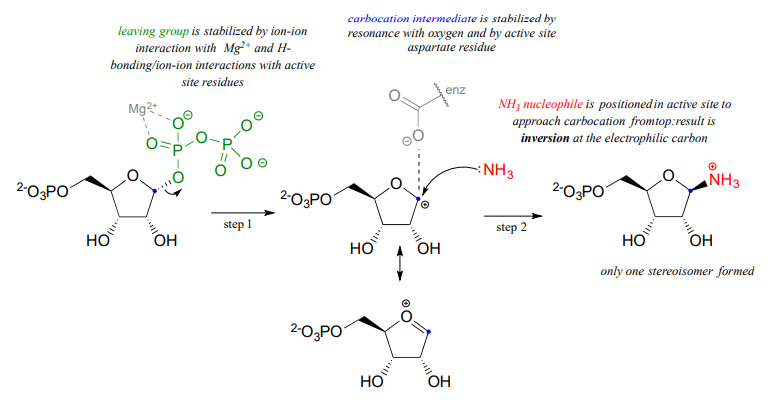
Observe algunas cosas aquí: primero, el grupo saliente difosfato se estabiliza por interacciones con\(Mg^{+2}\) iones unidos en el sitio activo y también por enlaces de hidrógeno con restos de aminoácidos del sitio activo (no mostrados). El carbocatión intermedio se estabiliza por resonancia con los pares solitarios en el oxígeno (ver sección 8.5), y también por una cadena lateral de aspartato de sitio activo. El nucleófilo de amoníaco se posiciona en el sitio activo de manera que se aproxima desde el lado 'superior' del intermedio de carbocatión plano, y la sustitución da como resultado inversión de configuración. Recuerde:\(S_N1\) las reacciones que ocurren libres en solución tienden a dar como resultado una mezcla de estereoisómeros, pero las reacciones catalizadas por enzimas -incluyendo\(S_N1\) reacciones enzimáticas como esta- son generalmente estereoespecíficas y regio-específicas, lo que significa que casi siempre dan como resultado un solo producto isomérico, no una mezcla de productos.
Recordemos la afirmación de la sección 8.4 de que los grupos pobres que salen a menudo necesitan ser convertidos en buenos grupos de salida. Respaldando un paso metabólico de la reacción descrita anteriormente, vemos que un grupo saliente pobre (hidróxido) en la ribosa-5-fosfato se convierte primero en un grupo saliente bueno (difosfato), que puede estabilizarse a través de interacciones con el sitio activo de la enzima que cataliza la\(S_N1\) reacción.
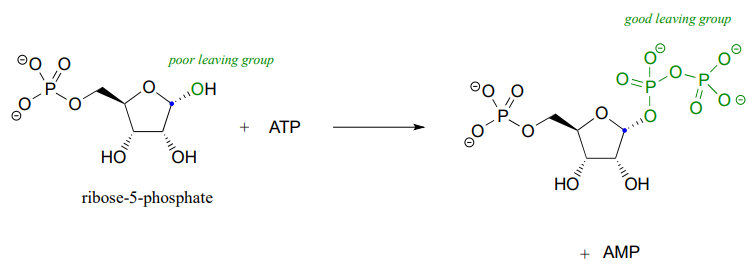
Esta etapa preliminar de fosforilación, que requiere ATP (trifosfato de adenosina) como donante del grupo difosfato, es una reacción que estudiaremos con mucho más detalle en el capítulo 9.
Una reacción\(S_N1/S_N2\) híbrida bioquímica
Los residuos de cisteína de ciertas proteínas se modifican mediante la adición de una cadena de isopreno de 15 carbonos (sección 1.3) al grupo tiol de la cadena lateral.
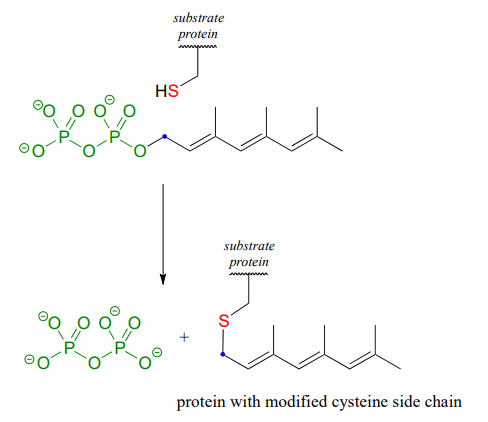
Los detalles mecanicistas de esta reacción son de particular interés para los científicos biomédicos. Las proteínas que son sustratos para este tipo de modificación están involucradas en los procesos de señalización celular, y no son capaces de llevar a cabo sus funciones biológicas a menos que estén ancladas a la membrana lipídica de una célula. El grupo hidrocarburo que se une a un residuo de cisteína en esta reacción sirve como anclaje.
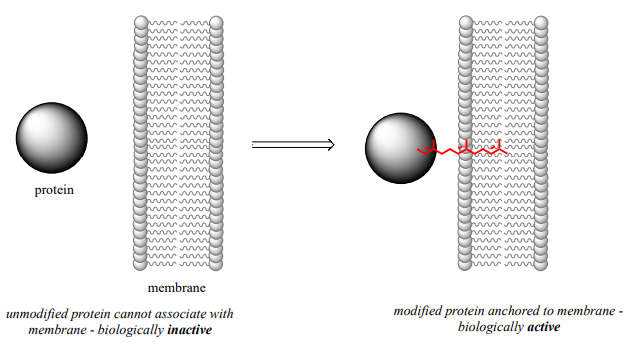
Algunas de estas proteínas han sido implicadas en la formación de tumores. Los científicos esperan que si pueden encontrar una manera de cerrar la reacción de modificación de cisteína, las proteínas causantes de tumores no podrán anclarse a las membranas celulares y así permanecerán inactivas. Se busca un inhibidor eficaz de esta enzima que sirva como posible fármaco antitumoral.
¿Cómo baja la enzima la barrera energética para esta reacción? La evidencia experimental indica que cuando una proteína sustrato se une al sitio activo de la enzima, el tiol de cisteína se asocia con un ion zinc unido en el sitio activo. Como aprendimos en la sección 7.8, esta asociación bajará el pKa del tiol hasta el punto en que pierda un protón y exista como anión tiolato en el sitio activo - ¡un tiolato es un nucleófilo muy potente! Los estudios también muestran que el grupo difosfato forma interacciones estabilizadoras con varios residuos de aminoácidos (dos lisinas, una arginina, una histidina y una tirosina) en el sitio activo de la enzima, convirtiéndola en una base más débil y, por lo tanto, un mejor grupo saliente.
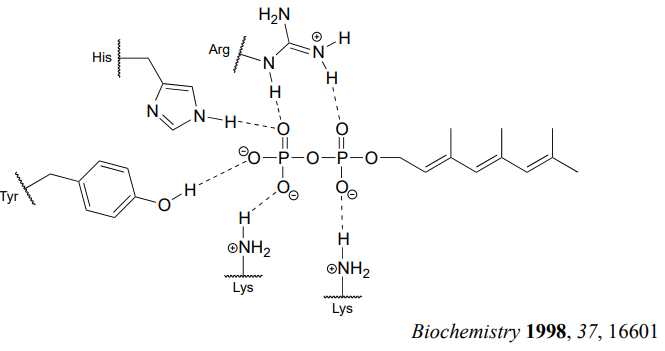
¿La prenilación de proteínas es una\(S_N2\) reacción\(S_N1\) o? En otras palabras, ¿hasta qué punto el nucleófilo desplaza, o 'empuja' al grupo saliente, o en qué medida el grupo saliente se va solo, sin un 'empuje' del nucleófilo? En la misma línea, en qué medida se desarrolla una carga positiva sobre el centro de carbono (el desarrollo de una carga positiva completa implica un\(S_N1\) mecanismo). Primero, considere el electrófilo: es un carbono alílico primario, por lo que cualquiera de las vías es posible (está relativamente libre de obstáculos para el\(S_N2\) ataque, pero también podría formar un carbocatión intermedio estabilizado por resonancia en una\(S_N1\) vía). El nucleófilo es un ion tiolato muy poderoso, sugestivo de un\(S_N2\) mecanismo donde un nucleófilo fuerte desplaza activamente al grupo saliente.
De hecho, experimentos diseñados para abordar esta misma cuestión (ver problema P8.19) han aportado evidencia de que la reacción es un híbrido mecanicista: esencialmente\(S_N2\), pero con elementos de\(S_N1\). Es decir, en el estado de transición el carbono electrófilo adquiere cierto grado de carga positiva, pero no se forma un verdadero intermedio de carbocatión. El mensaje para llevar a casa aquí es que las imágenes\(S_N1\) y\(S_N2\) mecanicistas que hemos estudiado en este capítulo son modelos, y si bien son útiles para aprender sobre principios químicos y precisas para describir muchas reacciones de sustitución, otras reacciones no son necesariamente 'puras'\(S_N1\) o \(S_N2\), pero en realidad se encuentran en algún punto intermedio.