II. Desoxigenación: La reacción de Barton-McCombie
- Page ID
- 80295
A. Una secuencia de dos pasos
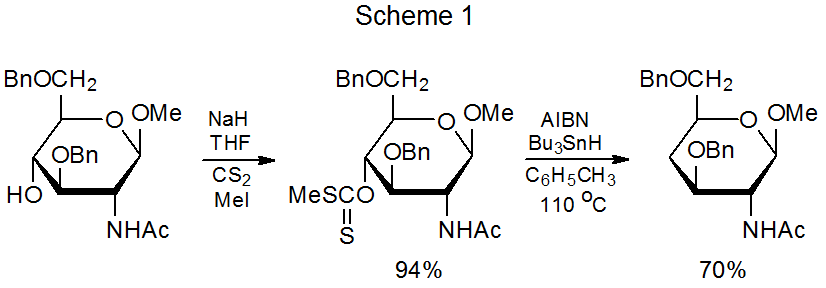
En 1975 Barton y McCombie reportaron una secuencia de dos etapas para el reemplazo del grupo hidroxilo por un átomo de hidrógeno. 1 El primer paso en este proceso fue la conversión del grupo hidroxilo en un grupo O-tiocarbonilo, y el segundo paso (la reacción de Barton-McCombie) fue una reacción en cadena de radicales libres que reemplazó al grupo O-tiocarbonilo con un átomo de hidrógeno. Un ejemplo típico de esta secuencia de reacción ampliamente utilizada se muestra en el Esquema 1. 2
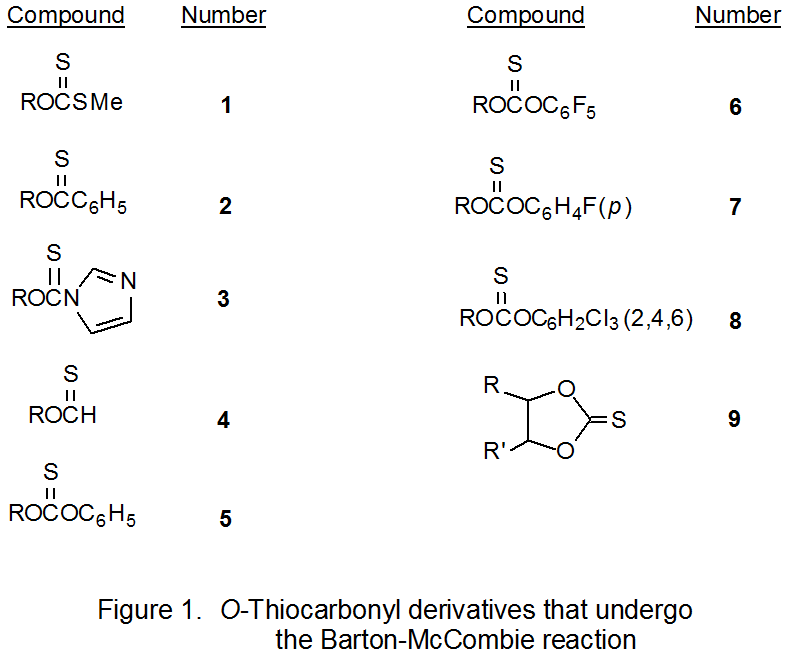
Varios tipos de compuestos de O- tiocarbonilo se someten a la reacción de Barton-McCombie. Inicialmente este grupo consistió en xantatos (1), tionobenzoatos (2), tiocarbonilimidazolidos (3) y tionoformiatos (4) (Figura 1). 1 Posteriormente, esta lista se amplió para contener tionocarbonatos de fenilo (5), 3,4 incluyendo aquellos con sustituyentes aceptores de electrones en el anillo aromático (6 - 8), 5,6 y tionocarbonatos cíclicos (9). 7,8
B. Mecanismos de reacción propuestos
Un mecanismo propuesto para la reacción de Barton-McCombie se muestra en el Esquema 2. 1,9 En la fase de inicio de esta reacción, la descomposición térmica de 2,2'-azobis (isobutirnitrilo) (AIBN) (eq 1), el iniciador más común para la reacción de Barton-McCombie, produce un radical que abstrae un átomo de hidrógeno del hidruro de tri- n-butilestaño (eq 2). En la primera etapa de propagación el radical tri- n-butilestaño se agrega a un doble enlace carbono-azufre para crear el radical aducto 10 (eq 3). La reacción alcanza una etapa crítica en este punto porque su éxito requiere 10 para fragmentar para dar el radical 11 (eq 4) antes de que puedan intervenir reacciones competitivas. Una vez que se produce la fragmentación, la abstracción de átomos de hidrógeno por 11 a partir de hidruro de tri- n-butilestaño completa la reacción global y genera un nuevo radical tri- n-butilestaño portador de cadena (eq 5). (Las ecuaciones 1-5 se encuentran en el Esquema 2.)
.png)
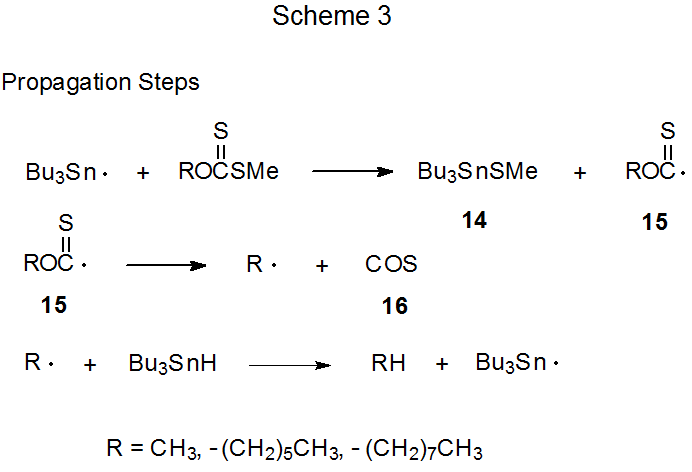
Los pasos de propagación para una propuesta mecanicista revisada para la reacción de Barton-McCombie se muestran en el Esquema 3. 10 (Los pasos de inicio y terminación para este mecanismo son los mismos que los que se muestran en el Esquema 2.) El cambio primario introducido en el mecanismo revisado (Esquema 3) es que Bu 3 Sn· no agrega al grupo tiocarbonilo sino que abstrae el grupo SCH 3. La identificación del radical 15 en el espectro ESR de la mezcla de reacción respalda el mecanismo revisado; sin embargo, un argumento en contra de la significación mecanicista de 15 es que este intermedio se observa en condiciones muy diferentes a las de la reacción de Barton-McCombie ( ej., en la mezcla de reacción no estuvo presente ningún donante efectivo de átomos de hidrógeno (Bu 3 SnH) debido a que los radicales tri- n-butilestaño se generaron a partir de la fotólisis de Bu 3 SnSnBu 3. 10)
Los experimentos de competencia posteriores devolvieron soporte al mecanismo original (Esquema 2). 11—13 Además de estos experimentos, la identificación por RMN de 119 Sn del intermedio 12 (R = SCH 3) en una mezcla de reacción Barton-McCombie proporcionó evidencia para la adición de Bu 3 Sn· al grupo tiocarbonilo, como se propone en Esquema 2, en lugar de abstracción de SCH 3, como se propone en el Esquema 3. 13 Otros análisis mecanicistas llevaron a la conclusión de que el radical tri - n-butilestaño debe estar agregando reversiblemente al grupo tiocarbonilo para dar el radical 10, que luego se fragmenta como se muestra en la ecuación 4 (Esquema 2). El soporte adicional para este mecanismo se basa en una reacción de formación de anillos que se describe al final de este capítulo, luego de que se hayan discutido las reacciones de ciclación.
C. Donantes de átomos de hidrógeno/agentes de transferencia de cadena
Crítico para el éxito de la reacción Barton-McCombie es el compuesto que dona un átomo de hidrógeno al radical carbohidrato (eq 5) para completar la secuencia de propagación. El hidruro de tri- n-butilestaño es particularmente adecuado para este papel porque dona rápidamente un átomo de hidrógeno a un radical centrado en carbono, y en la misma reacción genera el radical portador de cadena Bu 3 Sn·, un intermedio necesario para iniciar una nueva secuencia de etapas de propagación (eq 3). (Una característica crítica de la reactividad de Bu 3 Sn· es que no causa reacciones secundarias al extraer los átomos de hidrógeno de los enlaces carbono-hidrógeno).
Aunque el uso de hidruro de tri- n-butilestaño tiene ventajas significativas, también adolece de inconvenientes sustanciales. Existen serios problemas asociados con la toxicidad de los compuestos que contienen estaño y la dificultad para eliminar los residuos de estos compuestos de los productos de reacción. Se han propuesto diversas soluciones a estos problemas. Debido a que estas soluciones se aplican no sólo a los compuestos de O-tiocarbonilo sino también a una amplia gama de derivados de carbohidratos, no se discutirán aquí; más bien, se han reunido y se encuentran en el Apéndice I.
D. El alcance y la reactividad de los compuestos de O-tiocarbonilo
El número de derivados de carbohidrato de O-tiocarbonilo que se someten a la reacción de Barton-McCombie es grande y continúa creciendo. Aunque todos estos compuestos reaccionan básicamente de la misma manera, algunos son más adecuados que otros para situaciones particulares. Las siguientes secciones se centran en el alcance y la reactividad especial de diversos compuestos de O-tiocarbonilo. Para enfatizar su amplio rango de reactividad, se proporcionan referencias a la reacción de Barton-McCombie que tiene lugar en diversas posiciones en carbohidratos sustituidos. Estas referencias no pretenden representar el número total que existe, sino más bien proporcionar ejemplos de las reacciones posibles en carbohidratos cíclicos y de cadena abierta.
1. Xantatos
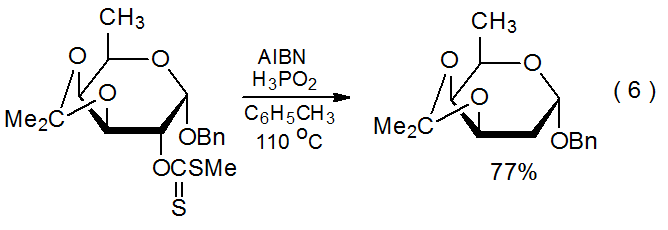
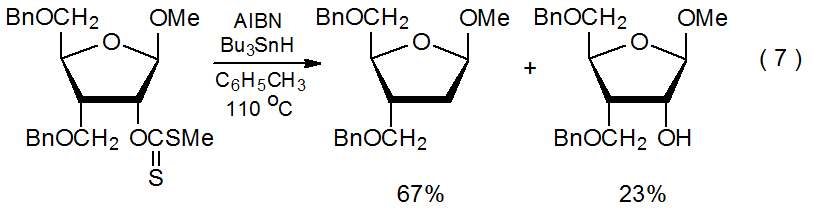
Una característica llamativa de la reactividad del xantato es la cantidad y variedad de carbohidratos que pueden desoxigenarse por la formación de xantato seguido de la reacción de Barton-McCombie. Esta secuencia reemplaza los grupos hidroxilo en las posiciones 2-, 14,15 3-, 16,17 4-, 2,18 y 6-19,20 con átomos de hidrógeno en compuestos que contienen anillos piranoides, así como las 1-, 21 2-, 22,23 3-, 24,25 5-, 26 y 6- 27 posiciones en aquellos con anillos furanoides. Las ecuaciones 6 15 y 7 28 proporcionan ejemplos típicos de reacciones de carbohidratos que contienen anillos piranoides y furanoides, respectivamente. (El ácido hipofosforoso, uno de los sustitutos del hidruro de tri- n-butilestaño mencionado en el Apéndice I, es el donador de átomos de hidrógeno en la reacción mostrada en la ecuación 6.) Los xantatos también se utilizan en la reacción Barton-McCombie de alditoles, 29,30 ciclitoles, 31,32 y nucleósidos. 33,34
.png)
.png)
2. Tionocarbamatos
Entre los tionocarbamatos los (tiocarbonil) imidazolidos (3, Figura 1) son fácilmente los sustratos más utilizados para la reacción de Barton-McCombie. El rango de tipos de compuestos involucrados es amplio e incluye (tiocarbonil) imidazolidos formados a partir de carbohidratos con grupos hidroxilo en las posiciones 2-, 35 3‑, 36—38 4-, 39—41 y 6-42 en compuestos con anillos piranoides, y en las 2‑, 43,44 3-, 45,46 y 5‑ 47,48 posiciones en compuestos con anillos furanoides. También hay numerosos reportes de reacciones Barton-McCombie de nucleósidos con grupos O‑ imidazol-1-iltiocarbonilo en C-2'‑ 49,50 y C-3'. 51,52
3. Tionocarbonatos
a. Tionocarbonatos de fenilo
Los tionocarbonatos de fenilo son otro derivado de O - tiocarbonilo, carbohidrato que se somete a la reacción de Barton-McCombie. Estos derivados participan en la reacción a C-2, 53,54 C-3, 55,56 C-4, 57,58 y C‑6 59,60 en compuestos con anillos piranoides, y C-1, 61,62 C-2, 63,64 C‑3, 65,66 y C‑5 67,68 en compuestos con anillos furanoides . Además, los tionocarbonatos de fenilo son los intermedios preferidos para la desoxigenación de nucleósidos. Están involucrados en la reacción en la posición 2' no solo para un gran número de nucleósidos protegidos con 1,1,3,3-tetraisopropil-1,3-disiloxanodiilo, 69,70 sino también para compuestos protegidos por bencilo, 71 benzoílo, 72,73 t-butildimetilsililo, 74 y pivaloílo grupos. 75 Reacciones similares tienen lugar en las posiciones 3' y 5'. 65,76
b. Fenil Tionocarbonatos sustituidos
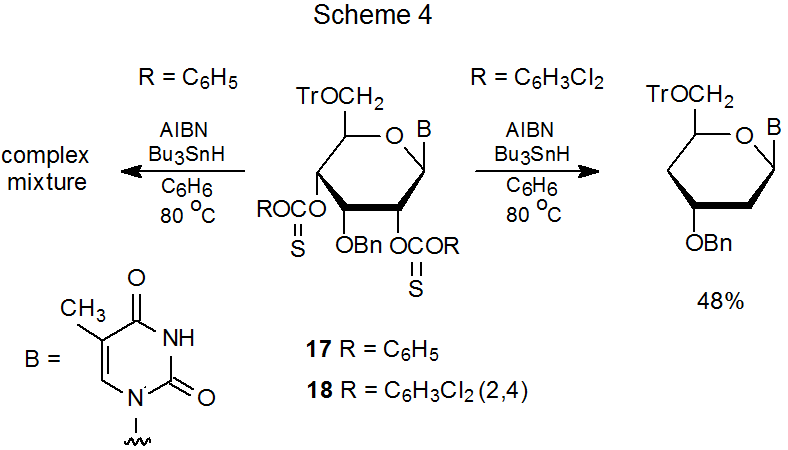
Los tionocarbonatos de fenilo típicamente experimentan una reacción de Barton-McCombie que produce productos desoxigenados con buen rendimiento; sin embargo, cuando los rendimientos del producto son bajos, la introducción de uno o más sustituyentes aceptores de electrones en el anillo aromático a menudo aumenta estos rendimientos. 77—80 Dos ejemplos ilustran el efecto de estos sustituyentes. Primero, el intento de reacción Barton-McCombie del fenil-tionocarbonato 17 produce una mezcla compleja de productos, pero el análogo 2,4-diclorofenilo 18 forma el didesoxi nucleósido deseado (Esquema 4). 77 En el segundo ejemplo, el tionocarbonato de fenilo 19 no es reactivo en condiciones típicas de Barton-McCombie, pero su análogo de pentafluorofenilo 20 reacciona con hidruro de tri- n-butilestaño para dar el desoxinonucleósido 21 correspondiente (eq 8). . 78
.png)
A la luz de los mejores rendimientos producidos por los fenil-tionocarbonatos sustituidos 18 y 20, es sorprendente encontrar que los derivados p-fluoro -, pentafluoro -, p-cloro -, 2,4,6-tricloro- y pentaclorofenoxitiocarbonilo del ciclododecanol todos reaccionan más lentamente que el compuesto no sustituido. 6 Al intentar comprender tal hallazgo, es útil considerar las diversas formas en que los sustituyentes del anillo pueden influir en la reactividad. La evidencia del estudio de compuestos de O-tiocarbonilo sugiere que la formación del radical 23 (eq 9) es un proceso reversible y que el paso determinante de la velocidad en esta secuencia de reacción (ecuaciones 9 y 10) es la fragmentación de 23 mostrada en la ecuación 10. 9 Es probable que un sustituyente de anillo aromático impacte en este proceso de varias maneras. Estos incluyen alterar la radicofilicidad de 22, la estabilidad de 23, y la fuerza del enlace carbono-oxígeno que se rompe para producir R· y 24 (eq 10); por lo tanto, dado que un sustituyente de anillo aromático puede ejercer influencia sobre la reactividad de varias maneras, comprender y predecir el efecto general que tal sustituyente tendrá en una reacción puede ser difícil.
.png)
c. Tionocarbonatos cíclicos
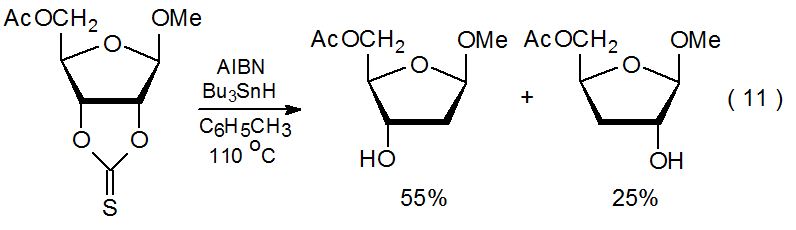
Los tionocarbonatos cíclicos sometidos a reacción de Barton-McCombie incluyen compuestos en los que los grupos 2,3-O-tiocarbonilo, 81—83 3,4-O-tiocarbonilo, 53,84 y 4,6-O-tiocarbonilo 85 están unidos a anillos piranoides. Esta reacción también se lleva a cabo con grupos 2,3- O-tiocarbonilo 7,8 y 5,6-O-tiocarbonilo 67,86 en compuestos con anillos furanoides. Los tionocarbonatos cíclicos, formados a partir de estructuras acíclicas, también experimentan la reacción de Barton-McCombie. 87
La reacción de un tionocarbonato cíclico es más compleja que la reacción de otros compuestos de O-tiocarbonilo porque también implica la apertura del anillo. Dado que la apertura del anillo potencialmente puede colocar un centro radical en cualquiera de los dos átomos de carbono, la reacción a menudo produce una mezcla de productos (eq. 11). 7,8 La formación de esta mezcla no sólo puede reducir la cantidad del producto deseado sino que también puede complicar su aislamiento.
.png)
Un mecanismo propuesto para la reacción de un tionocarbonato cíclico con hidruro de tri-n-butilestaño se da en el Esquema 5. 8 Un intermedio “clave” en esta reacción es el radical 25, formado por la adición del radical tri- n-butilestaño al grupo tiocarbonilo. La estabilidad radical generalmente controla la dirección de apertura del anillo; así, aunque 25 pueden producir un radical primario o uno secundario por apertura del anillo, la vía seguida conduce exclusivamente al radical secundario (Esquema 5). 7,8,88,89
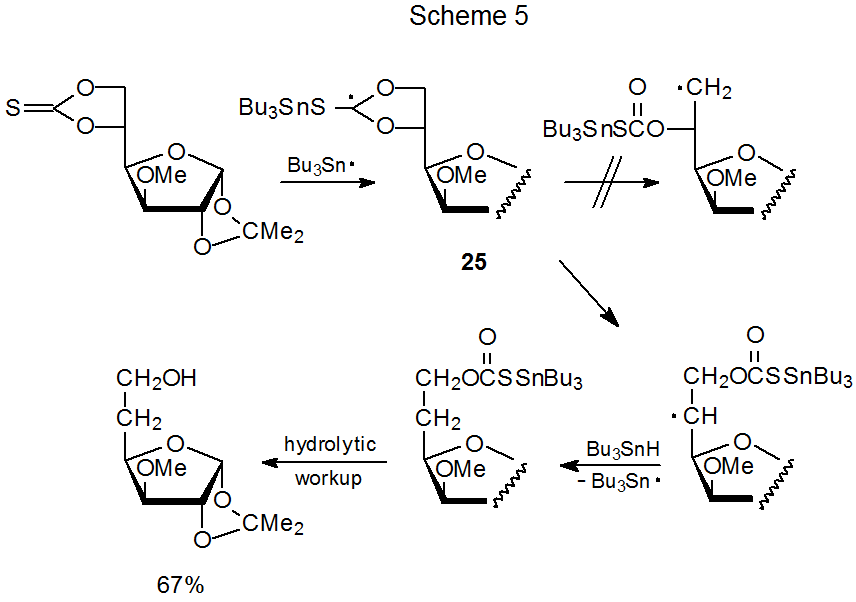
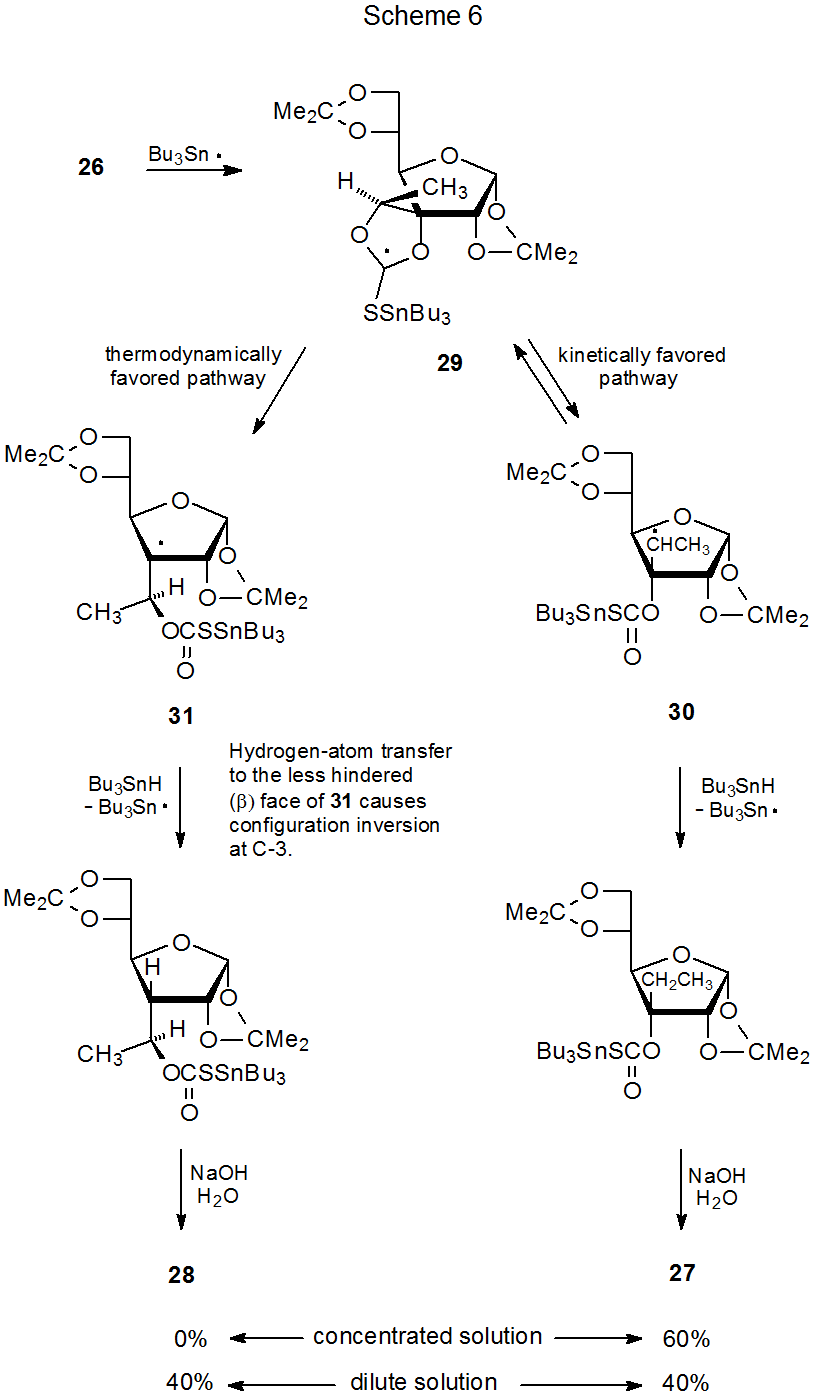
Aunque la estabilidad radical normalmente controla la dirección de apertura del anillo en un tionocarbonato cíclico, el alivio de la tensión angular en el estado de transición a veces es el factor principal. 90 La apertura del anillo del compuesto 26, por ejemplo, conduce al producto derivado de un radical secundario, en lugar de terciario (eq 12). 91 (Un mecanismo para esta reacción se muestra en el Esquema 6.) Los cálculos de la mecánica molecular en los no carbohidratos indican que la fragmentación para dar un radical menos estable ocurrirá si el alivio de la tensión del anillo en el estado de transición es lo suficientemente grande (eq 13). 90 Este alivio de la tensión proporciona una explicación para la inesperada conversión de 26 en 27 en lugar de 28 (eq 12).
.png)
.png)
Existe un efecto de concentración asociado a la reacción del tionocarbonato cíclico 26. En la solución concentrada de Bu 3 SnH solo se forma el producto cinéticamente favorecido 27, pero en solución diluida se produce parte del isómero termodinámicamente favorecido 28 (Esquema 6). Una explicación de este comportamiento se basa en la formación reversible del radical secundario 30 a partir del radical cíclico 29. En solución concentrada 30 extrae un átomo de hidrógeno lo suficientemente rápido de Bu 3 SnH para evitar un retorno significativo a 29. Bajo estas condiciones solo se forma el producto 27. En solución diluida, la abstracción de átomos de hidrógeno en 30 se ralentiza hasta el punto de que la formación reversible de 29 se vuelve significativa y crea una mayor oportunidad para que 29 se conviertan (irreversiblemente) en el radical terciario termodinámicamente favorecido 31. Dado que en solución diluida de Bu 3 SnH se siguen vías tanto cinética como termodinámicamente favorecidas, se produce una mezcla de los productos 27 y 28.
4. Tionoésteres
Principalmente debido a que su síntesis es más desafiante, los tionoésteres se seleccionan con menos frecuencia como materiales de partida para la reacción de Barton-McCombie que otros derivados de O-tiocarbonilo. Los tionoésteres con grupos O-tiocarbonilo en C-2 92 y C-3 1 en anillos piranoides y C-2 28 en anillos furanoides son sustratos conocidos para la formación de desoxi azúcar. Un ejemplo típico se muestra en la ecuación 14. 1 Los tionoésteres que se someten a reacción son, con raras excepciones, 93 tionobenzoatos. 28,92,94—101
.png)
E. Reacciones en competencia
La aplicación frecuente de la reacción Barton-McCombie en la química de carbohidratos ocurre porque esta reacción tiene una variedad de características atractivas. Estos incluyen la amplia gama de compuestos que experimentan esta reacción, los rendimientos generalmente buenos del producto y la libertad, en la mayoría de los casos, de reacciones significativas y competitivas. Aunque las reacciones secundarias generalmente no representan una preocupación importante, la reacción de Barton-McCombie se ha llevado a cabo en tantos compuestos que se han identificado bastantes de estas reacciones. Van en importancia desde la regeneración del alcohol, una reacción secundaria común pero generalmente menor, hasta la migración del grupo fenilo, un evento raro.
1. Regeneración de Alcohol
Regenerar el alcohol a partir del cual se sintetizó originalmente un compuesto de O-tiocarbonilo es una reacción secundaria que acompaña a veces la desoxigenación. Un ejemplo de tal reacción se muestra en la ecuación 15. 102 En raras ocasiones, la regeneración de alcohol es la principal vía de reacción (eq 16). 42
.png)
.png)
No todos los derivados de O - tiocarbonilo de los carbohidratos son igualmente propensos a la regeneración del alcohol. Una de las ventajas inicialmente asociadas con los tionocarbonatos de fenilo fue que no sufrieron esta reacción. 3,4 El estudio continuado de estos compuestos, sin embargo, demostró que no son inmunes al proceso de reforma del alcohol (eq 17). 103
.png)
a. Mecanismos de reacción propuestos
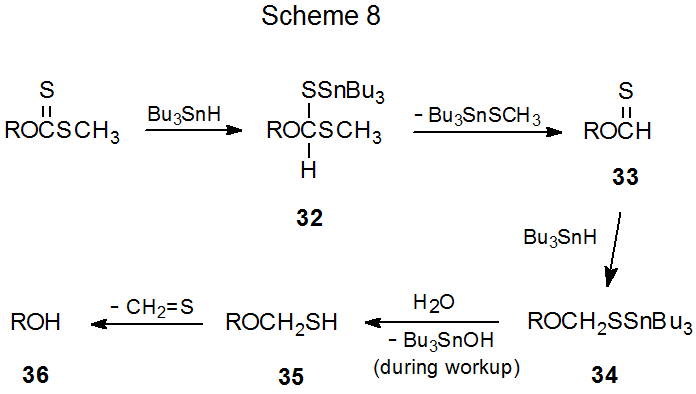
Aunque la regeneración de alcohol es el proceso de competencia más común durante la reacción de Barton-McCombie, no hay acuerdo general sobre cómo procede el proceso de regeneración. Las dos posibilidades más frecuentemente citadas son procesos de múltiples etapas con muchos intermedios en común. La diferencia entre estos dos descansa con la reducción del grupo tiocarbonilo, un mecanismo (Esquema 7) involucra intermedios radicales y el otro (Esquema 8) no.
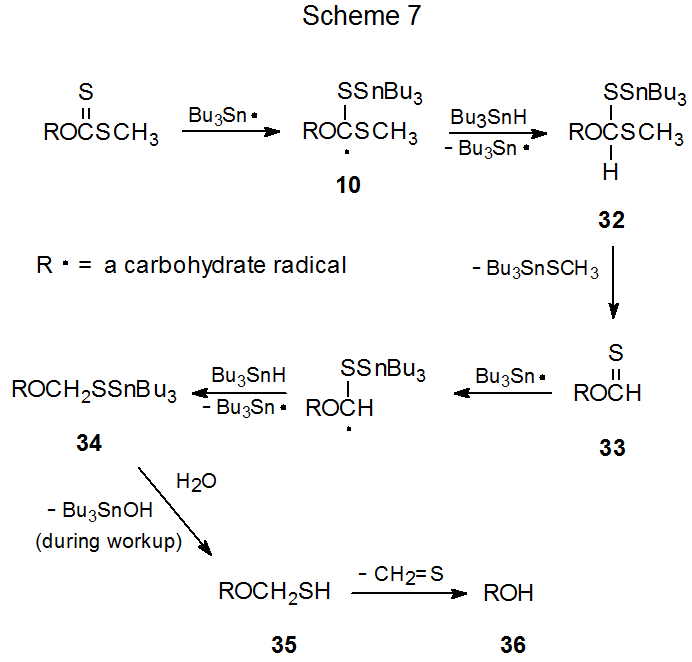
(1). Un Proceso Basado en Radicales
Si se asume que el radical 10 es un intermedio en la reacción de Barton-McCombie (Esquema 2, X = SCH 3), existe la posibilidad de que antes de que este radical se fragmente, pudiera abstraer un átomo de hidrógeno. Tal reacción produciría el intermedio 32 (Esquema 7). 1,9 La formación de 32 no solo reduciría el rendimiento del producto desoxigenado, sino que también podría proporcionar una explicación para la formación del alcohol regenerado 36 como producto secundario en la reacción. 1,9 Cuando se ve de esta manera, 32 es el punto inicial de una serie de eventos que conducen primero al tionoformiato 33, que reacciona con hidruro de tri- n - butilestaño para dar el intermedio 34 que contiene estaño, una sustancia que se hidroliza durante tratamiento al hemitioacetal 35. El compuesto 35 luego se descompone espontáneamente para dar el alcohol 36 y tioformaldehído (Esquema 7). 9
(2). Una secuencia de reacción basada en transferencia de hidruro
Las condiciones originalmente utilizadas para la reacción de Barton-McCombie no incluyeron un iniciador agregado; más bien, la reacción dependió de la iniciación adventicia. En pocos años, sin embargo, la adición de 2,2'-azobis (isobutirnitrilo) (AIBN) se convirtió en un procedimiento estándar debido a que la iniciación confiable se reconoció como un factor significativo para maximizar la desoxigenación. 4,13 La reacción mostrada en la ecuación 18 es una que proporciona una ilustración de la mejora en el rendimiento del producto producida por un iniciador añadido. 104 La observación de que la regeneración de alcohol ocurre en ausencia de un iniciador en la reacción mostrada en la ecuación 18, respalda la idea de que una reacción radical puede no estar involucrada en el proceso de reforma del alcohol. 4
.png)
Cuando la reacción Barton-McCombie es iniciada por Et 3 B—O 2, 105,106 puede tener lugar a temperaturas muy inferiores a las requeridas para el inicio de AIBN (eq 19). 21 Debido a que originalmente se pensó que reducir la temperatura de una reacción de Barton-McCombie a aproximadamente 80 o C provocó que la regeneración del alcohol comenzara a ser importante, 12 se esperaría que una disminución adicional de la temperatura de reacción aumentara la formación de alcohol. Cuando las reacciones se realizaron a menor temperatura mediante el inicio de Et 3 B—O 2, la reacción de Barton-McCombie tuvo lugar con poca o ninguna regeneración de alcohol. Este hallazgo no fue consistente con la idea de que 10 (Esquema 7) era cada vez más probable que abstraviera un átomo de hidrógeno de Bu 3 SnH a medida que disminuía la temperatura de reacción. 13 Si la conversión de 10 en 32 por abstracción de átomos de hidrógeno no se realizaba, planteaba la posibilidad de que 32 se formara en una reacción diferente, quizás no radical, (Esquema 8).
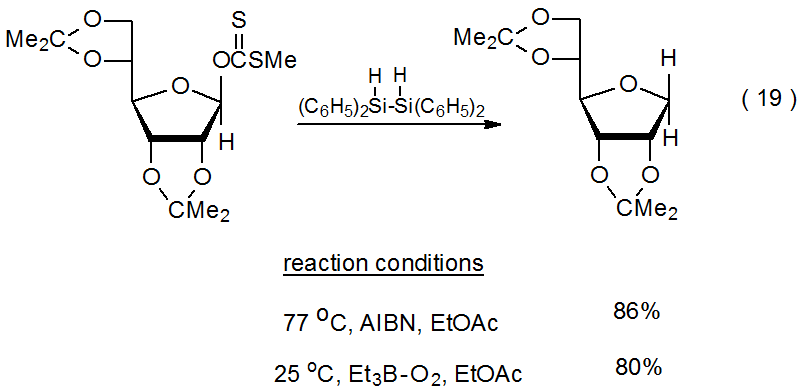.png)
Varios investigadores han propuesto que la transferencia de hidruro puede ser responsable de la regeneración del alcohol. 13,107,108 Como parte de la más detallada de estas propuestas, se sugirió que la adición no radical de hidruro de tri- n-butilestaño a un grupo O-tiocarbonilo ocurre en la primera etapa en la regeneración de alcohol, y se produce posteriormente en la secuencia de reacción cuando el el tionoformiato 33 se convierte en el tioacetal 34 (Esquema 8). 108
b. Alcoholes de Ésteres Diméricos
La observación de que se formaron pequeñas cantidades de ésteres “diméricos” durante la síntesis de tionocarbonato de fenilo mostrada en la ecuación 20 planteó la posibilidad de que si estos ésteres no se eliminaban antes de la reacción de Barton-McCombie, se podría formar un alcohol porque se podía esperar razonablemente que cada éster dimérico reaccionar con hidruro de tri- n-butilestaño para dar una molécula de un compuesto desoxigenado y uno de los alcoholes de partida. 103 Esta expectativa se confirmó cuando se encontró que el éster dimérico 37 experimentó la reacción mostrada en la eq 21. 109
.png)
.png)
c. Minimizar la regeneración de alcohol
Se cree que la etapa determinante de la velocidad en la reacción de Barton-McCombie es la fragmentación unimolecular del radical 10 para dar el radical centrado en carbono R· (Esquema 9). 9 Para la regeneración de alcohol, sin embargo, es más probable que la velocidad dependa de una reacción bimolecular que involucre Bu 3 SnH. Este análisis (Esquema 9) es consistente con el hallazgo de que mantener la concentración de Bu 3 SnH a un nivel bajo durante la reacción (es decir, agregar el hidruro de estaño lentamente a medida que avanza la reacción) se asocia con maximizar la desoxigenación; por ejemplo, el xantato que se muestra en la ecuación 22 reacciona para dar un desoxi-azúcar con 54% de rendimiento cuando todo el Bu 3 SnH está presente al inicio de la reacción, pero el rendimiento aumenta a 85% cuando se agrega Bu 3 SnH durante un periodo de 1.5 h. 36 Parte del rendimiento de desoxi-azúcar reducido en la reacción donde se añadió todo el Bu 3 SnH en el inicio se debe a la recuperación del alcohol a partir del cual se sintetizó el xantato. 36
.png)
2. Conversiones y reordenamientos del grupo O-tionocarbonilo
a. Conversión de un Xantato en un Ditiocarbonato
Cuando Bu 3 SnH es el donante de átomos de hidrógeno en la reacción mostrada en eq 23, 1 se forma un desoxi azúcar de la manera normal, pero si se usa un donante menos efectivo, la conversión de xantato a un ditiocarbonato compite con el proceso de desoxigenación. Esta conversión se vuelve significativa cuando la reacción se lleva a cabo con 2-propanol que sirve como disolvente y donante de átomos de hidrógeno. Cuando el benceno es el disolvente, la formación de ditiocarbonato es la vía de reacción principal. 110 El benceno es, de hecho, un donante de átomos de hidrógeno tan pobre que cualquier carbohidrato presente en solución es una fuente de átomos de hidrógeno más probable para la pequeña cantidad de desoxi azúcar formada.
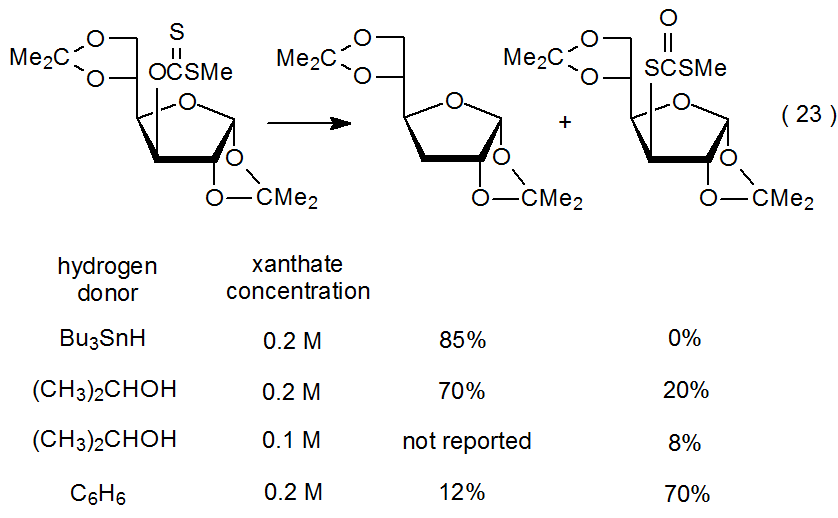.png)
La reacción mostrada en la ecuación 23 comienza con la formación del radical carbohidrato R·. Como se muestra en el Esquema 10, este radical (R·) luego se agrega a una molécula de material de partida, lo que conduce a un ditiocarbonato, o abstrae un átomo de hidrógeno, produciendo un desoxi azúcar. Los datos presentados en la ecuación 23 confirman la expectativa del mecanismo propuesto (Esquema 10) de que la formación de desoxi-azúcar se ve favorecida cuando se utilizan donantes efectivos de átomos de hidrógeno, y los ditiocarbonatos se forman más fácilmente en reacciones ejecutadas a altas concentraciones de xantato en presencia de donantes pobres de átomos de hidrógeno . 110
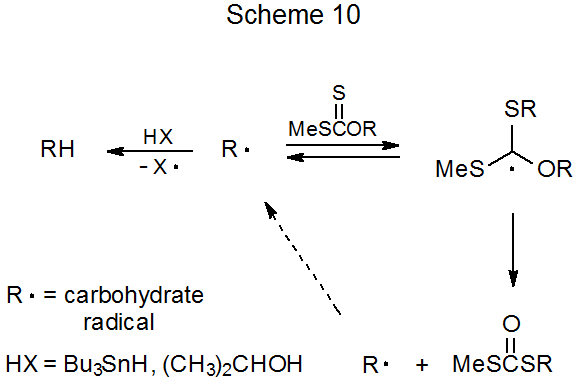
b. Reordenamiento de Tionocarbonato-Tiocarbonato
Una reacción estrechamente relacionada con el reordenamiento de xantato-ditiocarbonato que se acaba de discutir es la conversión de un tionocarbonato en un tiocarbonato. Cuando un donante de átomos de hidrógeno está presente en una mezcla de reacción en una cantidad menor que la necesaria para suministrar un átomo de hidrógeno a cada radical carbohidrato, puede tener lugar el reordenamiento de tionocarbonato a tiocarbonato. 82,88,89,111,112 En la reacción mostrada en el Esquema 11, este reordenamiento representa la vía principal de reacción cuando la cantidad de hidruro de tri- n-butilestaño es significativamente menor que la requerida para la reducción completa. 111 El reordenamiento es, por supuesto, indeseable cuando la simple reducción es el objetivo de una reacción, pero puede ser útil si el propósito de la reacción es convertir un azúcar en un tioazúcar. 112
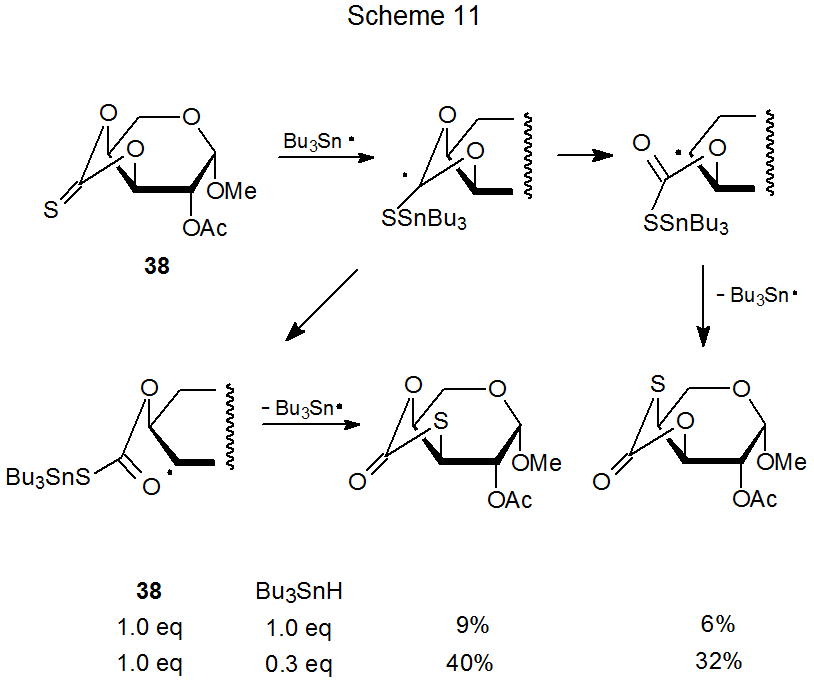
c. Conversión de un Xantato de S-alquilo en un Xantato de O-alquilo
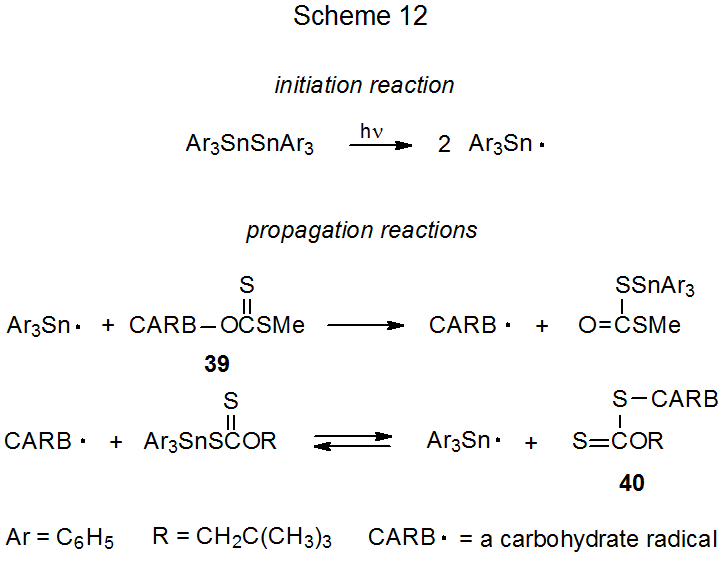
La reacción del carbohidrato xantato 39 (un S-alquil xantato) con el radical trifenilestaño es el primer paso en una secuencia de propagación (Esquema 12) que convierte 39 en un xantato con la porción carbohidrato de la molécula unida al azufre (40, un O -alquil xantato). 113,114 El segundo paso en esta secuencia es la reacción del radical carbohidrato con xantato de trifenilestaño para producir 40 y el radical trifenilestaño portador de cadena. Esta segunda etapa debe ser reversible para dar cuenta de que los epímeros 41 y 42 se interconvierten bajo las condiciones de reacción (eq 24). 114 Los donantes efectivos de átomos de hidrógeno, como el hidruro de trifenilestaño, deben excluirse para evitar una reducción simple. Excluyendo hidruro de trifenilestaño pero teniendo aún el radical trifenilestaño necesario para iniciar la reacción se logra por fotólisis de bis (trifenilestaño) (Esquema 12).
.png)
3. Reacción con Oxígeno Molecular
La reacción realizada en presencia de oxígeno molecular conduce a una rápida captura de O 2 por radicales centrados en carbono. Esta captura es más rápida que la abstracción de átomos de hidrógeno de Bu 3 SnH. La captura radical de O 2 se suprime mediante el procedimiento normal de excluir el oxígeno de la mezcla de reacción, pero cuando se agrega deliberadamente O 2, su combinación con un radical carbohidrato se convierte en una ruta de reacción principal o incluso exclusiva (eq 25). 89
.png)
4. Reacciones de eliminación
a. Reacciones de Compuestos con Dos Grupos O-Tiocarbonilo
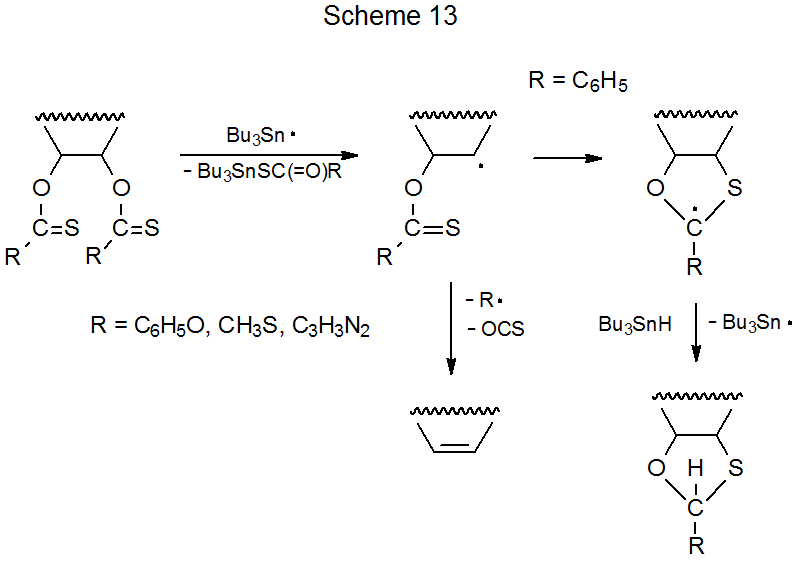
Si hay dos grupos O- tiocarbonilo en la misma molécula, su separación física afecta si se llevará a cabo o no la reacción de Barton-McCombie. 1,115—134 En la reacción del compuesto 43, por ejemplo, los sustituyentes están suficientemente bien separados para permitir la sustitución de cada grupo por un átomo de hidrógeno para proceder de la manera normal (eq 26 115). 115,130 Cuando los grupos O- tiocarbonilo están unidos a átomos de carbono adyacentes, la reacción de uno de estos grupos produce un radical centrado en carbono que forma un doble enlace mediante la eliminación del segundo grupo. 117—129,131,133 Un ejemplo se muestra en la ecuación 27. 117—119 Como se indica en el Esquema 13, la eliminación se realiza cuando R = OC 6 H 5 o SCH 3, pero en el raro caso de que R = C 6 H 5, la vía de eliminación conduce al radical fenilo inestable; como consecuencia, la ciclación radical se lleva a cabo en lugar de eliminación (eq 28). 1 Si dos grupos O-tiocarbonilo no están en átomos de carbono adyacentes, pero el radical producido por reacción del primero está centrado en un átomo muy próximo al segundo, se llevará a cabo la adición interna. 115,116,132,134 Debido a que el nuevo radical formado por esta reacción no tiene un camino claro hacia un producto de eliminación, es probable que tenga lugar una reacción más compleja, como la que se muestra en la ecuación 29, 115.
.png)
.png)
.png)
.png)
b. Reacciones de Compuestos con un Solo Grupo O-Tiocarbonilo
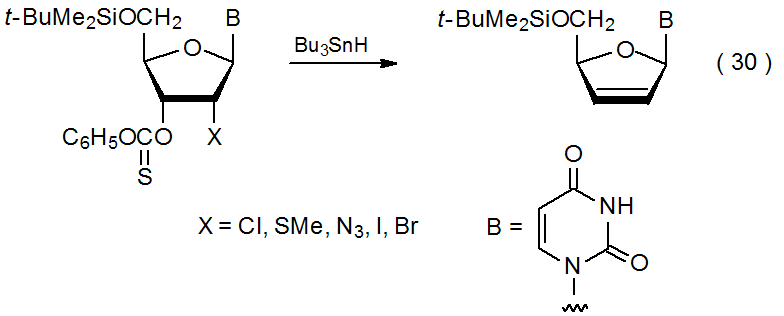
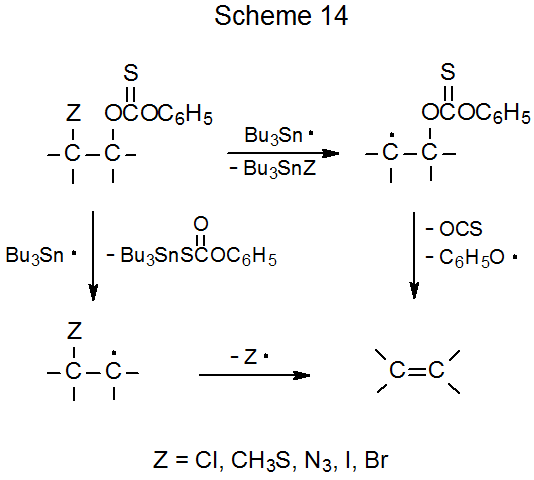
Como se describe en la sección anterior, las reacciones de eliminación tienen lugar cuando los compuestos con grupos O-tiocarbonilo en átomos de carbono adyacentes reaccionan con hidruros de estaño o silicio. Reacción similar tiene lugar cuando un grupo O-tiocarbonilo está unido a un átomo de carbono que tiene un azido, 135 bromo, 135—137 cloro, 135,138,139 iodo, 135 isóciano, 140 metiltio, 135 o feniltio, 141 sustituyente unido a un átomo de carbono adyacente. Un ejemplo se da en la eq 30. 135 Este proceso comienza con la formación de un radical centrado en carbono y termina con la expulsión radical de un átomo de carbono adyacente (Esquema 14). En algunos casos es la reacción del grupo O- tiocarbonilo la que genera el radical centrado en carbono, pero en otros, particularmente aquellos que involucran compuestos que contienen bromo y yodo, la abstracción de átomos de halógenos es la primera reacción que tiene lugar.
.png)
c. Uso de Olefinas Sacrificiales
Una de las dificultades asociadas con la síntesis de compuestos insaturados por reacción radical es que un intermedio radical puede agregarse a un doble enlace recientemente formado en una molécula producto. Cuando se produce tal adición no deseada, se puede reducir en importancia a un nivel aceptable añadiendo un alqueno tal como 1-dodeceno a la mezcla de reacción. Este alqueno, un compuesto descrito como una “olefina sacrificial”, protege el producto de eliminación al eliminar radicales antes de que se agreguen a las moléculas del producto. 142
d. Tionocarbonatos cíclicos
Una forma de ver los tionocarbonatos cíclicos es como compuestos que tienen grupos O-tiocarbonilo adyacentes y, en consecuencia, podrían sufrir eliminación radical. Dado que las reacciones Barton-McCombie de los tionocarbonatos cíclicos generalmente dan buenos rendimientos de compuestos desoxi, solo se puede esperar una reacción de eliminación si dicha reacción se beneficia de una fuerza motriz especial. Esta fuerza impulsora puede provenir de la reacción que produce un compuesto insaturado con un doble enlace lo suficientemente estable como para que su formación disminuya significativamente la energía del estado de transición. Consistente con esta idea es la formación del glical 46 como único producto de la reacción del tionocarbonato cíclico 45 con hidruro de tri- n-butilestaño (eq 31). 143 Los derivados de nucleósidos 2',3'- O-tiocarbonilo experimentan una reacción similar para dar nucleósidos insaturados, pero solo como productos menores. 144—146
.png)
e. Prevención de la eliminación térmica
La reacción Barton-McCombie de xantatos es más exitosa cuando estos compuestos se preparan a partir de alcoholes secundarios. Los xantatos formados a partir de alcoholes terciarios son propensos a la eliminación térmica (eq 32, eliminación Chugaev 147) a temperaturas normalmente utilizadas en la reacción de Barton-McCombie. Cuando la reacción es iniciada por trietilborón-oxígeno, sin embargo, se puede llevar a cabo a temperatura ambiente, donde la reacción de xantatos terciarios ocurre sin eliminación competitiva. 106,148
.png)
5. Adición reversible a un grupo O-tiocarbonilo
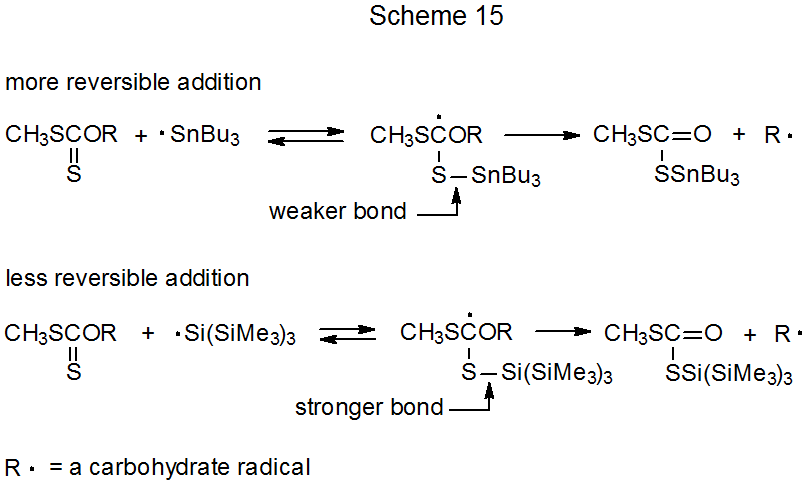
Los xantatos sintetizados a partir de alcoholes primarios generalmente requieren temperaturas más altas en la reacción de Barton-McCombie y a menudo dan rendimientos de producto más bajos. 149,150 Estos problemas pueden superarse en algunos casos cambiando el donante de átomos de hidrógeno de Bu 3 SnH a (Me 3 Si) 3 SiH. El hidruro de tri- n-butilestaño es un donador menos efectivo que el tris (trimetilsilil) silano en estas reacciones debido a la mayor reversibilidad de la adición de Bu 3 Sn· a un grupo tiocarbonilo (Esquema 15). 149 Debido a que el enlace S-Si [90 kcal mol -1 (377 kJ mol -1)] 151 es más fuerte que el enlace S-Sn [65 kcal mol -1 (272 kJ mol ‑1)], 151 inversión de la adición de (Me 3 Si) 3 Si· es menos probable que ocurrir. Reversibilidad reducida significa que una vez que un radical sililo se ha añadido a un grupo O-tiocarbonilo, una reacción directa difícil (por ejemplo, una que produce un radical primario 149 y, a veces, una que produce un radical secundario 152, 153) puede competir más eficazmente con la inversa. reacción para dar los materiales de partida. La limitación de la reversibilidad es aún más efectiva cuando la reacción tiene lugar en las condiciones de baja temperatura posibilitadas por el inicio de Et 3 B—O 2. 33
6. Reducción de un grupo tiocarbonilo a un grupo metileno
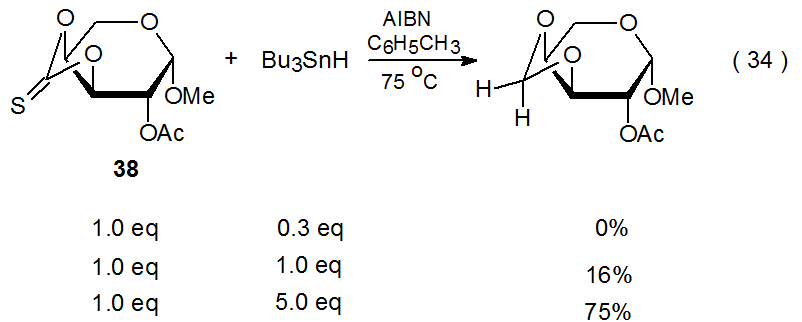
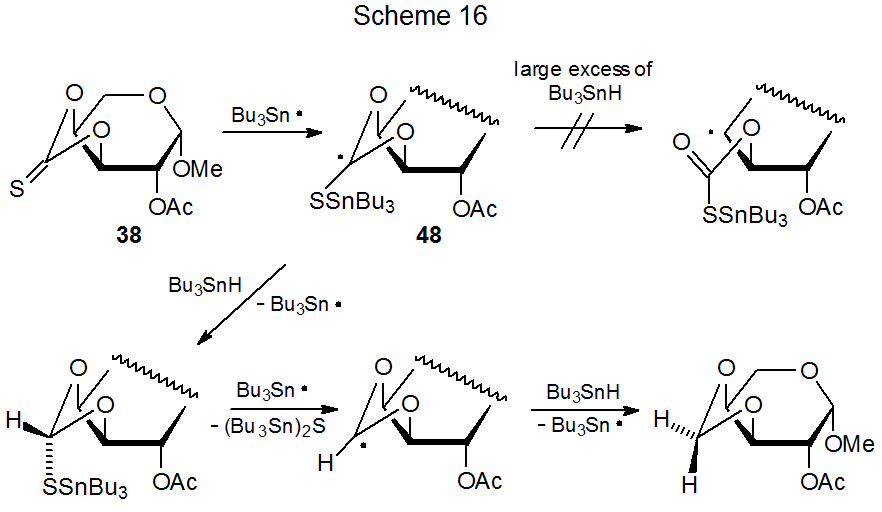
La conversión de un grupo tiocarbonilo en un grupo metileno representa un tipo raro de competencia para la reacción de Barton-McCombie. Esta transformación cambia el grupo O- feniltiocarbonilo en 47 en un grupo O-bencilo (eq 33), 28 y es responsable de un cambio similar en el tionocarbonato cíclico 38 (eq 34). 89,111 La reacción mostrada en la ecuación 34 ocurre con un rendimiento mucho mayor cuando está presente un gran exceso de hidruro de tri- n-butilestaño. Un efecto de un gran exceso de Bu 3 SnH es aumentar la tasa de abstracción de átomos de hidrógeno por el radical 48 (Esquema 16) hasta el punto de que la apertura del anillo por este radical es demasiado lenta para ser detectada.
.png)
.png)
7. Migración de grupos fenilo
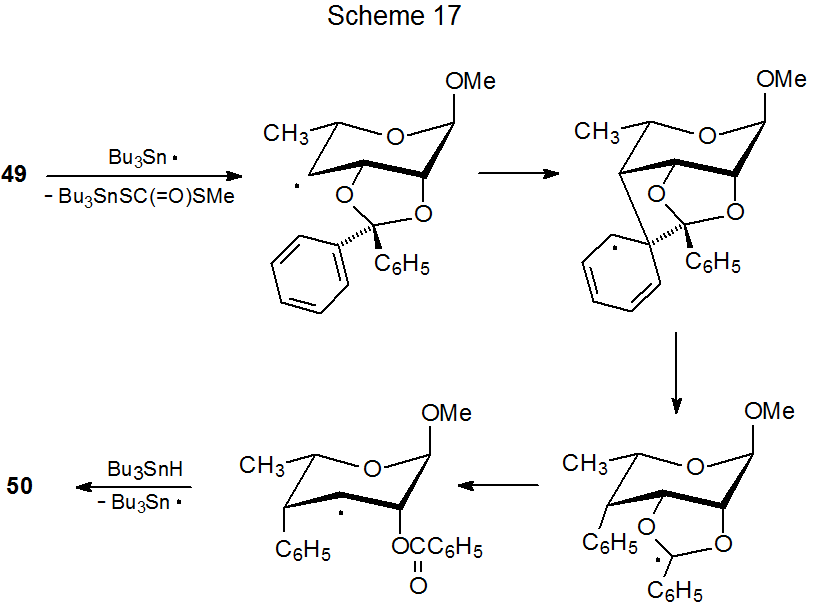
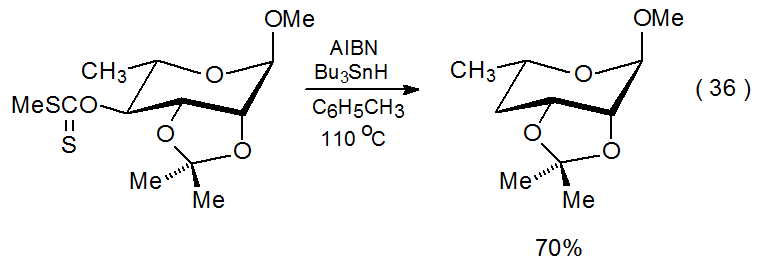
El compuesto 49, un xantato con un grupo O - [(alquiltio) tiocarbonilo] en la protección C-4 y 2,3-O-difenilmetileno, experimenta migración del grupo fenilo y apertura del anillo durante la reacción (eq 35). 154 De acuerdo con el mecanismo propuesto en el Esquema 17, esta reacción depende de la presencia de sustituyentes en el grupo protector que pueden sufrir migración por un proceso de adición-eliminación. Consistente con esta propuesta mecanicista es la observación de que si el grupo 2,3-O-difenilmetileno es reemplazado por un grupo 2,3-O-isopropilideno, se produce una reacción normal (eq 36). 155
.png)
.png)
F. Influencia de los efectos estéricos sobre la reactividad
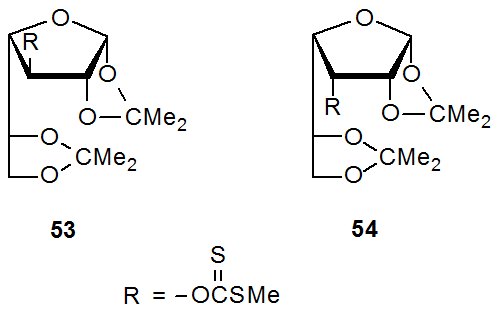
La reacción de los tionocarbamatos epiméricos 51 y 52 con Bu 3 SnH ilustra el papel que juegan los efectos estéricos en la reacción de Barton-McCombie (eq 37). 156 El ambiente estérico dramáticamente diferente para los sustituyentes C-3 en 51 y 52 no tiene un impacto sustancial en su reactividad; específicamente, la diferencia de un factor de cuatro en los tiempos de reacción (no se midieron las velocidades de reacción) es consistente con solo influencia menor por efectos estéricos. Una posible explicación de esta modesta diferencia de reactividad es que aunque la congestión estérica alrededor de C-3 en los compuestos 51 y 52 es sustancialmente diferente, no es dramáticamente diferente en el área del átomo de azufre, donde se está produciendo la reacción. La comparación de la reactividad de los xantatos 53 y 54 proporciona otro ejemplo del modesto papel de los efectos estéricos en la reacción de Barton-McCombie. El grupo O - [(metiltio) tiocarbonilo] decididamente más impedido en 54 hace que este xantato (54) solo unas cinco veces menos reactivo que su epímero 53. 157
.png)
G. Regioselectividad
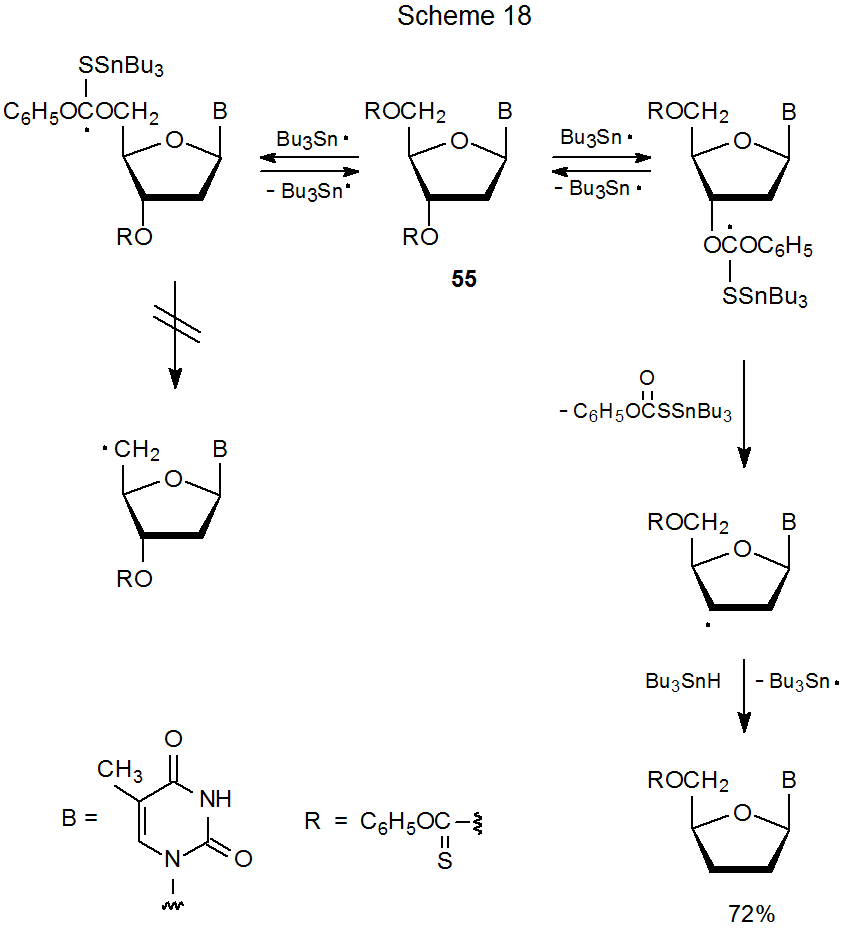
Una de las características de la reacción de Barton-McCombie es que cuando se realiza de manera normal (es decir, con Bu 3 SnH como donante de átomos de hidrógeno y AIBN como iniciador), se requiere una temperatura más alta para la reacción de un grupo O-fenoxitiocarbonilo primario que un secundario uno. 76,158 Esta diferencia en la reactividad puede convertirse en la base de la reacción regioselectiva; así, como se muestra en el Esquema 18, el reemplazo de grupos se lleva a cabo solo en la posición 3' en el nucleósido 55. 76
H. Quimioselectividad
La quimioselectividad en reacciones de compuestos que contienen grupos O-tiocarbonilo se discute en la Sección II.B del Capítulo 9 del Volumen I.
I. El Efecto β-Oxígeno
Una observación sobre la reacción de Barton-McCombie es que algunos carbohidratos reaccionan más fácilmente de lo que se esperaría sobre la base del comportamiento del compuesto modelo. Esta mayor reactividad se observa cuando el átomo de carbono en un enlace C-O está relacionado con β con un átomo de carbono que porta un grupo O-tiocarbonilo. 93 La comparación de las reactividades de 56 y 57 ilustra esta diferencia. El compuesto 56 no reacciona con hidruro de tri- n-butilestaño a 110 o C (lo hace a 130 o C), 93 pero 57, que también es una (tiocarbonil) imidazolida primaria, reacciona en estas condiciones 42,93 Este y similares obser llevaron a la propuesta de que “el oxígeno unido al β-carbono de un radical de carbono tiene un marcado efecto estabilizador; esto permite reacciones radicales no vistas, excepto a temperaturas mucho más altas, en compuestos modelo no oxigenados”. 93 Cuando se propuso por primera vez, este “efecto β-oxígeno” representó un factor potencialmente importante en la química de carbohidratos porque muchos radicales centrados en carbono serían estabilizados por su existencia.
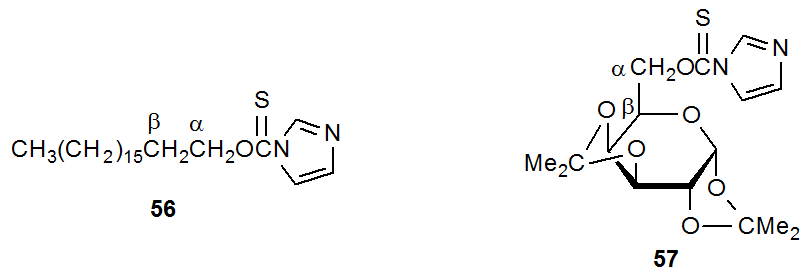
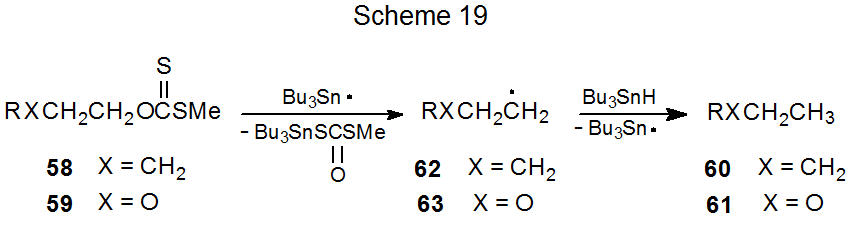
Se ha invertido un esfuerzo considerable en el estudio del efecto β-oxígeno. Este trabajo ha establecido que la presencia de un enlace carbono-oxígeno relacionado con β no necesariamente aumenta la tasa de formación de un radical en desarrollo centrado en carbono. Por ejemplo, la reacción de una mezcla de los xantatos 58 y 59 con una cantidad limitada de hidruro de tri- n-butilestaño dio los productos 60 y 61 en cantidades aproximadamente iguales (Esquema 19). 159,160 Dado que este resultado significó que los xantatos 58 y 59 reaccionaban esencialmente a la misma velocidad, un enlace carbono-oxígeno relacionado con β estaba proporcionando poca, si alguna, estabilización del estado de transición para el radical en desarrollo 63. 160
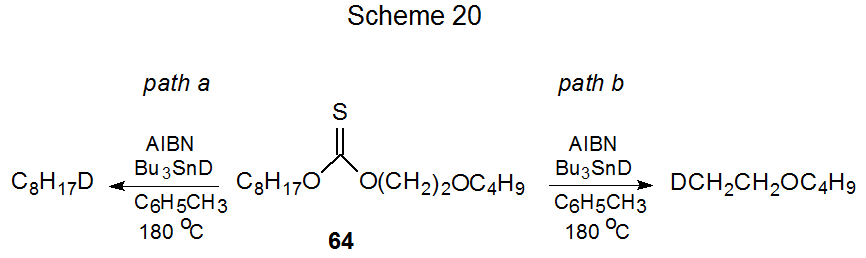
Un experimento relacionado involucró el tionocarbonato 64, el cual podría reaccionar a lo largo de cualquiera de las dos vías competidoras (Esquema 20). Si existiera el efecto β-oxígeno, se favorecería la reacción por vía b. La experimentación mostró que la reacción a lo largo de cada vía era igualmente probable; en consecuencia, la conclusión nuevamente fue que no existía evidencia de un efecto β-oxígeno. 149
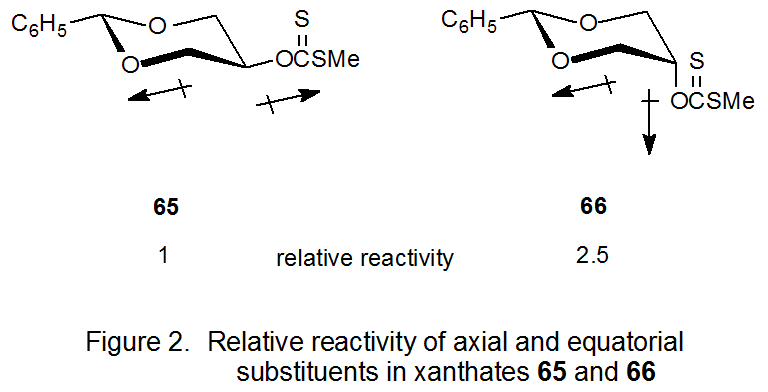
Aunque estos experimentos disiparon la idea de que un átomo de oxígeno relacionado con β generalmente proporciona estabilización a un centro radical, no explicaron por qué el derivado de carbohidrato 57 es más reactivo que el compuesto modelo 56. Sin embargo, una explicación de este comportamiento proviene del estudio de los isómeros ecuatorial y axial 65 y 66, respectivamente. El epímero axial (66) con sus dipolos gauche (sinclinales) es más reactivo que el epímero ecuatorial (65) con dipolos trans (Figura 2). La mayor reactividad del compuesto 66 se atribuye a la eliminación de una interacción dipolo-dipolo desfavorable durante la ruptura del enlace. La eliminación de tal interacción parece ser entonces la razón principal de las diferencias de velocidad que originalmente se atribuyeron al efecto β-oxígeno. 149