
II. Oximas
- Page ID
- 80367
A. Reacciones de adición
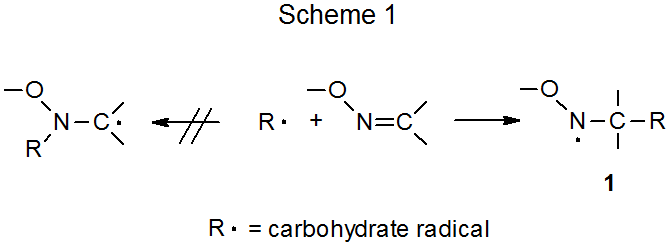
Un radical centrado en carbono se agrega preferentemente al átomo de carbono en el doble enlace carbono-nitrógeno de un éter de oxima (Esquema 1). La regioselectividad de esta reacción es consistente con un radical que se agrega al éter de oxima para producir el más estable de los dos posibles radicales aductos; es decir, el radical estabilizado por un átomo de oxígeno unido al centro radical. Ejemplos de este tipo de reacción se encuentran en la adición de un radical piranos-1-ilo a O-bencilformaldoxima (eq 1), 3 y la reacción de un radical nucleósido centrado en C-3' a un éter de oxima que contiene nucleósidos (eq 2). 4,5
.png)
.png)
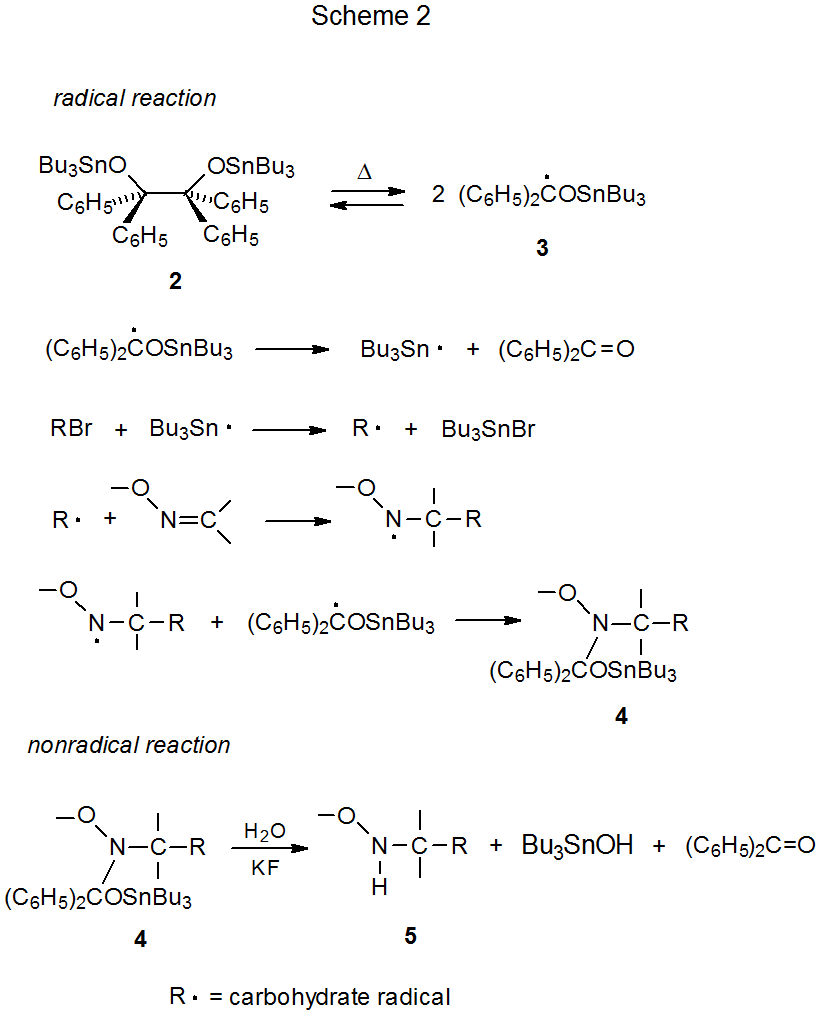
Los radicales centrados en carbono que se suman a los éteres de oxima se forman típicamente a partir de los yoduros y bromuros correspondientes por reacción con Bu 3 Sn·. Debido a que la reducción simple ofrece una competencia significativa a la adición radical, Bu 3 SnH no es la mejor fuente para los radicales centrados en el estaño que participan en estas reacciones. La adición es más exitosa cuando Bu 3 Sn· se genera a partir de bis (tri- n-butilestaño) benzpinacolato (2), un compuesto que produce Bu 3 Sn· pero suprime la reducción simple al no introducir una transferencia efectiva de átomos de hidrógeno en la mezcla de reacción (Esquema 2) . 3 En ausencia de tal donante, se piensa que la fase radical de esta reacción produce el compuesto 4 que contiene estaño, el cual luego se hidroliza a la benciloxiamina 5 en una reacción no radical (Esquema 2).
B. Reacciones de ciclación
1. Regioselectividad
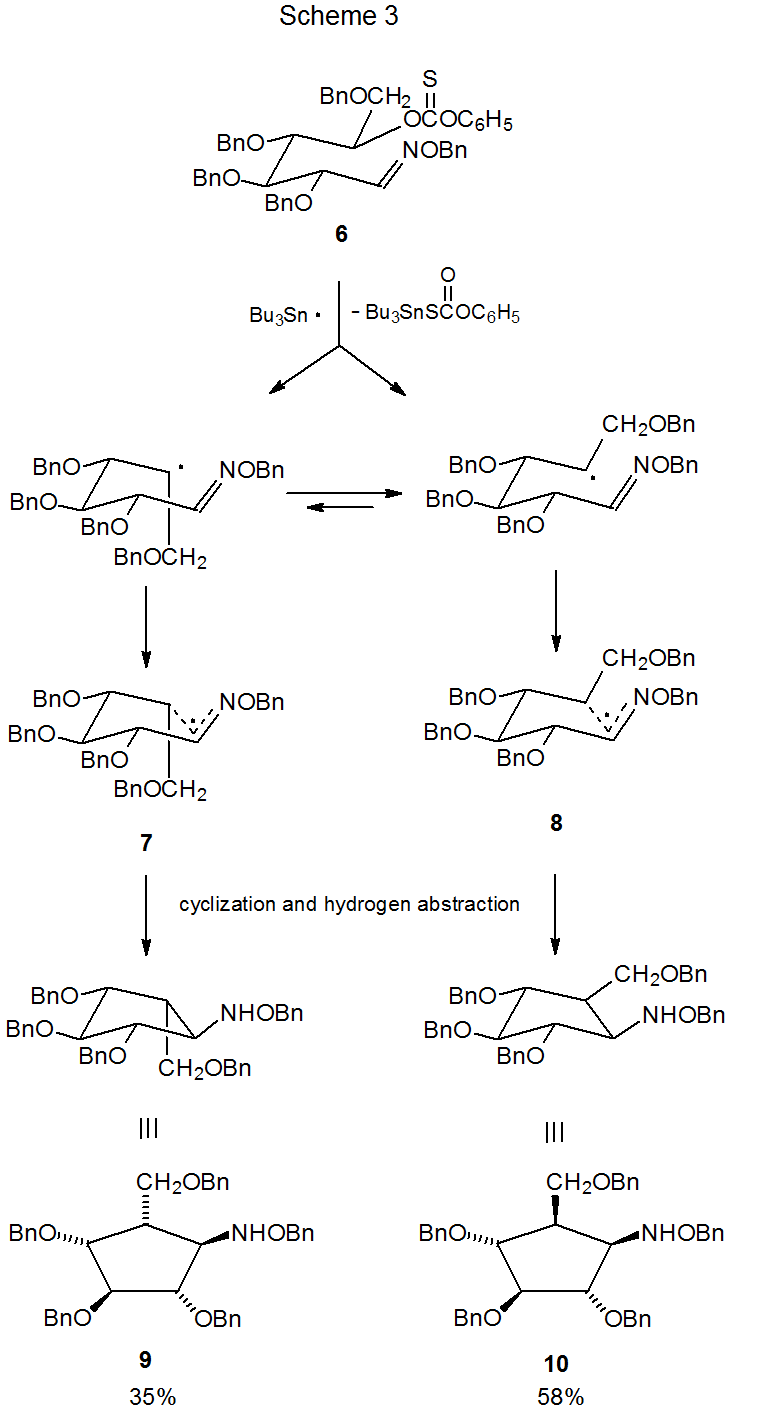
Un ejemplo de ciclación regioselectiva de radicales que implica un éter de oxima se muestra en el Esquema 3. 6 Un factor importante para determinar la regioselectividad en este tipo de reacción es la adición energéticamente favorecida de un radical al átomo de carbono del doble enlace carbono-nitrógeno (Esquema 1). También es importante la preferencia durante la ciclación radical de éteres de oxima para formar anillos de cinco miembros en lugar de seis miembros cuando ambos son posibles y anillos de seis miembros en lugar de siete miembros cuando cualquiera de ellos podría formarse. 7
2. Estereoselectividad
La ciclación radical convierte los éteres de oxima de cadena abierta en ciclopentanos 6 ,8—14 y ciclohexanos altamente sustituidos. 7,15—17 Como suele ser el caso en las reacciones de ciclación radical, se puede entender la estereoselectividad, si se asume que el estado de transición de energía más bajo es una estructura similar a una silla que maximiza el número de sustituyentes pseudoecuatoriales. La reacción mostrada en el Esquema 3, por ejemplo, genera compuestos con dos nuevos centros quirales pero produce solo dos (9 y 10) de los cuatro estereoisómeros posibles. 6 Estos dos isómeros surgen de los estados de transición tipo silla 7 y 8, respectivamente. El estado de transición 8, que conduce al producto principal 10 tiene el mayor número de sustituyentes pseudoecuatoriales.
A la luz de la reacción del éter de oxima 6 (Esquema 3) se podría esperar que el éter de oxima estructuralmente similar 11 (eq 3) también produzca solo los dos estereoisómeros 14 y 15, es decir, los productos esperados de la reacción a través de estados de transición tipo silla. En realidad, 11 produce los cuatro estereoisómeros posibles (eq 3), y ni 14 ni 15 es el producto principal. 11 El producto principal (12) surge de la reacción que involucra el estado de transición similar a la embarcación 16 (Esquema 4). A menudo se requieren cantidades similares de energía para alcanzar estados de transición similares a sillas y barcos en las reacciones de ciclación radical (Sección IV.A en el Capítulo 11 del Volumen I); en consecuencia, las diferencias en la estructura cerca de los centros reactivos pueden afectar significativamente la distribución del producto.
.png)
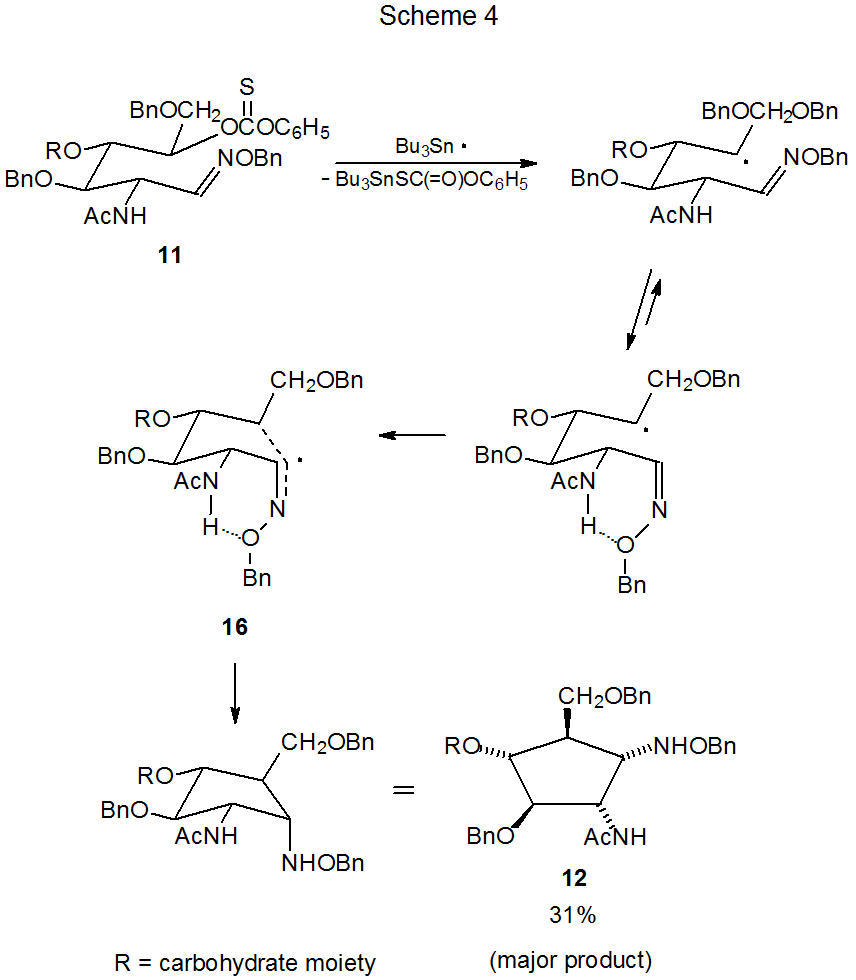
Una diferencia entre los sustratos 6 y 11 es que 6 tiene un grupo bencilooxi en C-2 (Esquema 3), y 11 tiene un grupo acetamido en esta posición (Esquema 4). Existe la posibilidad de que los enlaces de hidrógeno que involucran al grupo acetamido estabilice el estado de transición 16 hasta el punto de que la vía de reacción que contiene esta estructura tipo embarcación (16) se convierta en la de menor energía (Esquema 4).
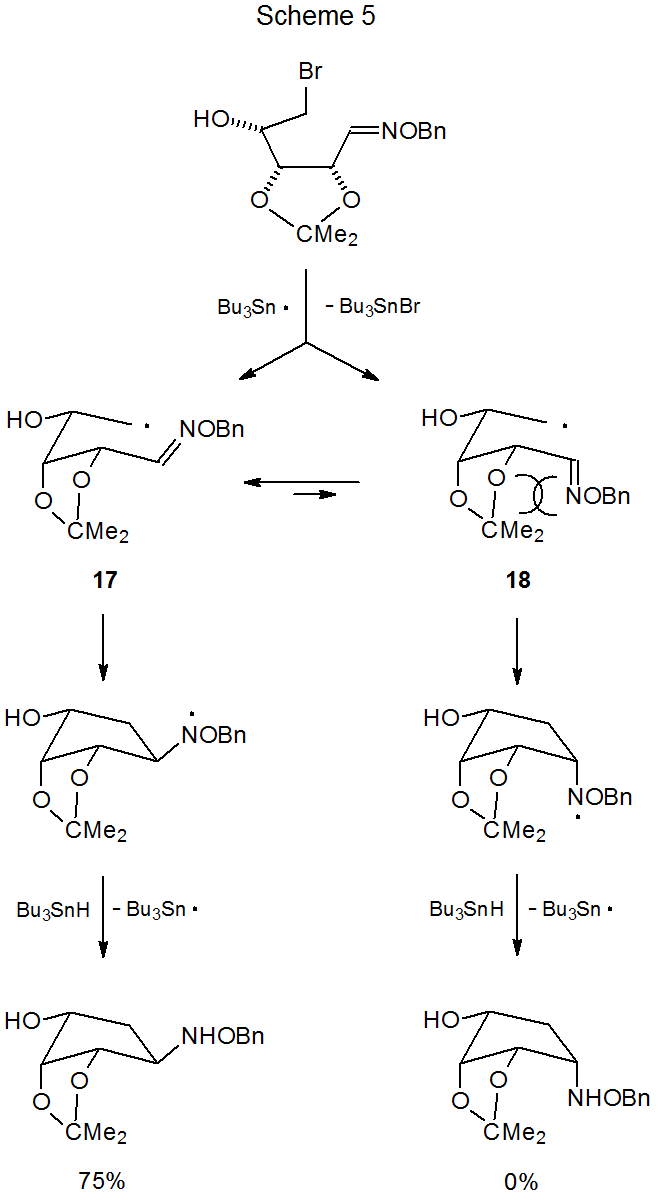
Un grupo O-isopropilideno que está cerca de los centros reactivos en una reacción de ciclación tiene un pronunciado efecto de estereoselectividad. 17—24 Este efecto se ilustra por la reacción mostrada en el Esquema 5, donde el único producto tiene el sustituyente NHOBn en el lado del anillo recién formado que es opuesto al grupo O-isopropilideno. 18 En esta reacción, la conformación tipo silla (17) del radical intermedio se ve muy favorecida sobre su contraparte similar a una embarcación (18) debido, en gran medida, a interacciones estéricas desestabilizadoras que involucran al grupo O-isopropilideno.
3. Reacciones formadoras de radicales
Los radicales de carbohidratos que se agregan internamente a los dobles enlaces carbono-nitrógeno provienen de una variedad de fuentes. Tales radicales a menudo se forman por reacción de compuestos de O-tiocarbonilo 6,8—11,20,21,23 o bromuros 15—19 con hidruro de tri- n - butilestaño. Las reacciones de Bu 3 SnH con compuestos carbonílicos 12 y ditioacetales 13 representan dos formas menos comunes para generar radicales que luego experimentan formación de anillos. Los radicales necesarios también se pueden formar por reacción de transferencia de electrones.
4. Preservar el Doble Enlace
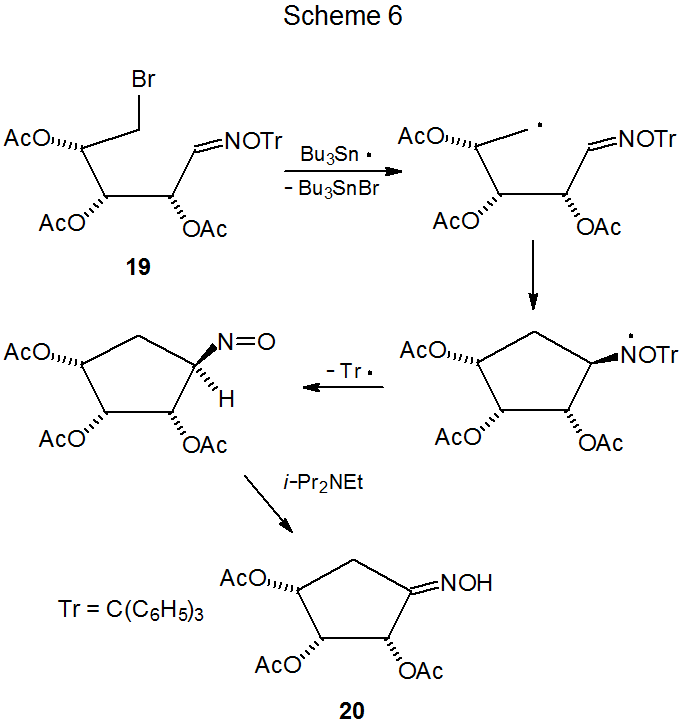
Normalmente, el doble enlace en un éter de oxima se convierte en un enlace sencillo durante la formación del anillo, pero cuando una oxima de O-tritilo se somete a ciclación, el doble enlace carbono-nitrógeno se conserva en el producto. Un mecanismo propuesto para este tipo de reacción se muestra en el Esquema 6, y los reactivos preferidos, todos los cuales son críticos para el éxito de la reacción, se dan en la ecuación 4. 24 La adición lenta de Bu 3 SnH y el iniciador ABC también son necesarias para la reacción sintéticamente útil. El radical persistente (C 6 H 5) 3 C· dificulta la propagación de la cadena por su lenta reacción con Bu 3 SnH; en consecuencia, para evitar la acumulación de (C 6 H 5) 3 C·, difenildiselenuro, un precursor conveniente para el hidrógeno muy reactivo- transferencia de átomos C 6 H 5 SeH, se puede agregar a la mezcla de reacción. Esta adición reemplaza una reacción lenta (eq 5) por dos rápidas (eq 6 y eq 7). 24 Si bien los carbohidratos que se han estudiado hasta ahora dan buenos rendimientos de producto sin adición de diselenuro de difenilo, otros compuestos no lo hacen; en consecuencia, la inclusión de selenuro en la mezcla de reacción ahora forma parte del procedimiento recomendado.
.png)
.png)
.png)
.png)
5. Reactividad mejorada de los radicales vinílicos
El intento de ciclación del radical generado a partir del bromuro 21 produce solo el producto de reducción simple 22 (eq 8), pero la reacción del bromuro vinílico relacionado 23 da tanto un producto cíclico como un producto de reducción simple (eq 9). 25 Un factor que puede contribuir a la diferencia en la capacidad de los compuestos 21 y 23 para someterse a ciclación radical es la considerable reactividad de los radicales vinílicos. Esta reactividad puede ser suficiente para provocar la formación de nuevo anillo durante la reacción de 23.
.png)
.png)
Además de la abstracción de átomos de halógeno, los radicales vinílicos también se forman por reacción de un radical centrado en estaño con un compuesto que contiene un triple enlace carbono-carbono. 26—30 Cuando un radical formado de esta manera se agrega internamente a un doble enlace carbono-nitrógeno, produce un producto cíclico que contiene estaño (eq 10). 26 El triple enlace en tales reacciones suele ser terminal, pero los enlaces internos, particularmente aquellos en conjugación con grupos estabilizantes de radicales, también experimentan reacción. 30
.png)
C. Reacción de transferencia de electrones
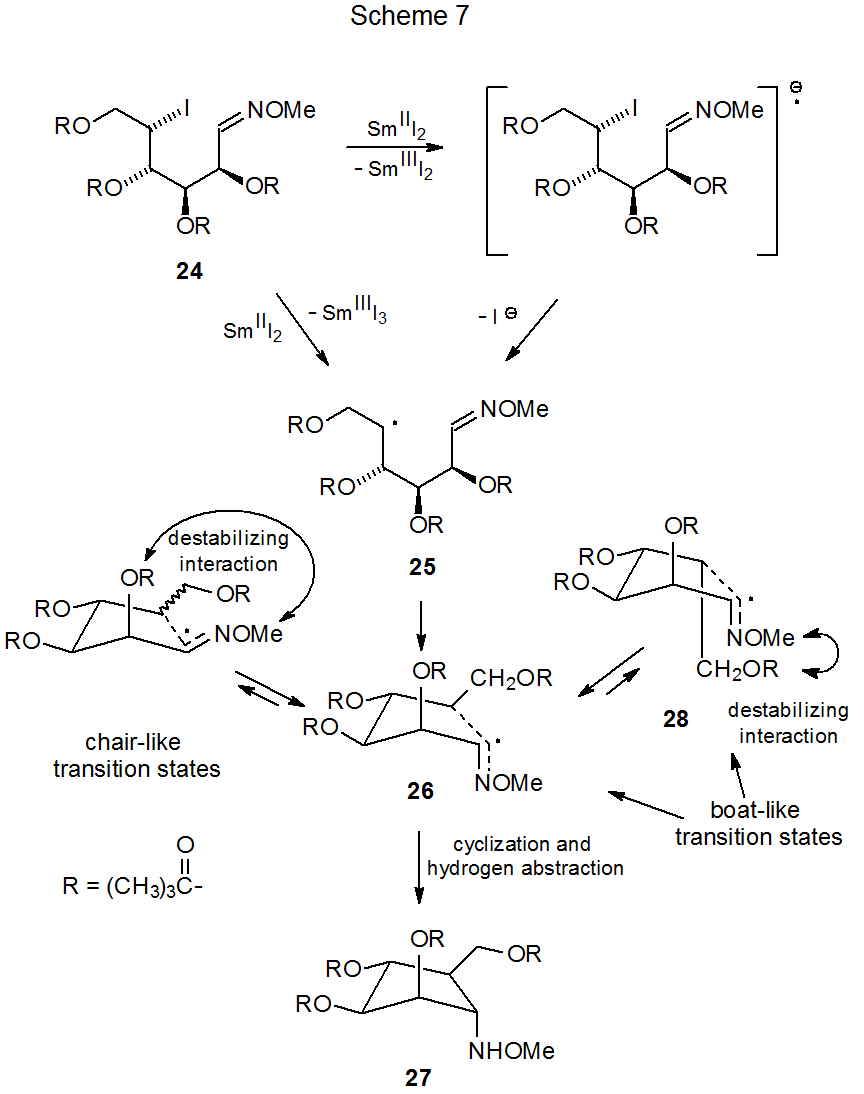
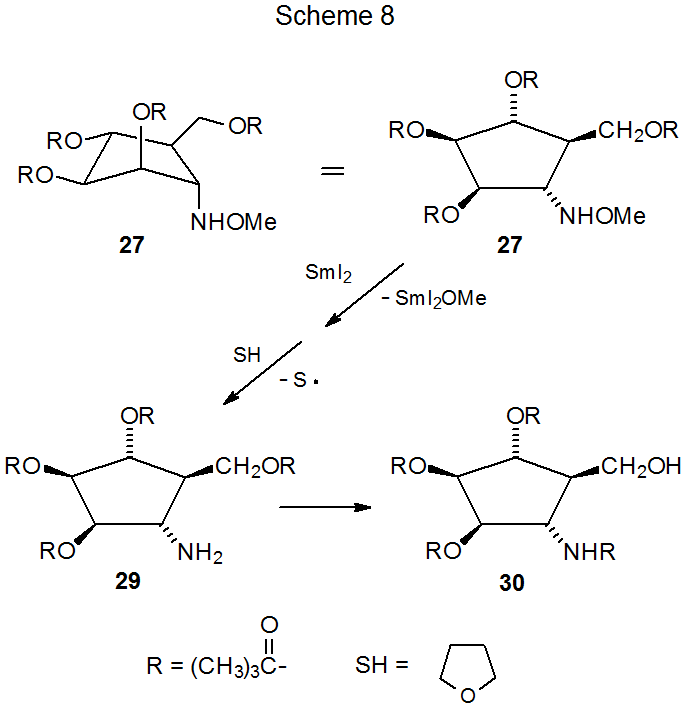
La transferencia de electrones proporciona otro medio para generar un radical formador de anillo a partir de un éter de oxima. El donador de electrones en tales reacciones es típicamente yoduro de samario (II) y el aceptor de electrones a menudo es un éter de oxima que contiene un átomo de halógeno. Una reacción típica se muestra en el Esquema 7, 31 donde la transferencia de electrones disociativa que involucra el enlace carbono-yodo en 24 conduce a la formación del radical 25 centrado en carbono. La ciclación y la abstracción de átomos de hidrógeno completan esta reacción formando el derivado de ciclopentilamina 27 en un proceso que pasa por el estado de transición tipo bote 26. Aunque los estados de transición tipo silla son típicamente favorecidos en las reacciones de ciclación, en este caso las estructuras tipo silla tienen interacción desestabilizadora entre los sustituyentes C-1 y C-2; además, el otro estado de transición tipo bote (28) tiene estabilidad reducida debido a la interacción entre los C Sustituyentes -1 y C-6 (Esquema 7). Si el exceso de SMi 2 está presente durante la reacción, la transferencia de electrones disociativa rompe el enlace N-O en 27 para producir, después de la abstracción del átomo de hidrógeno, la ciclopentilamina 29 correspondiente (Esquema 8). La migración no radical del grupo acilo del oxígeno al nitrógeno forma entonces el producto final (30). 31 (El yoduro de samario (II) también escinde los enlaces N-O que no se forman a partir de la reacción de éteres de oxima. 32)
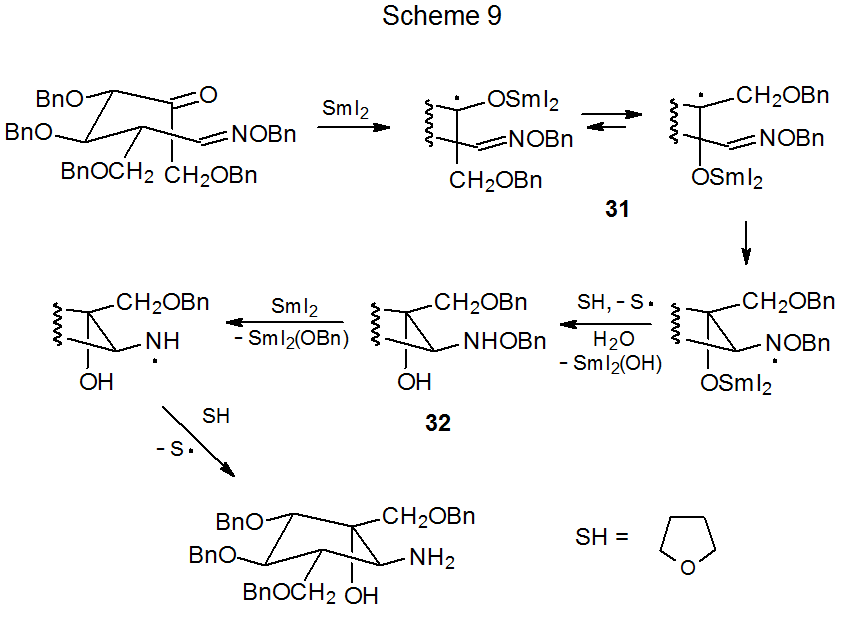
La transferencia de electrones a un grupo aldehido o ceto en un éter de oxima también puede ser la etapa inicial en la formación del anillo. 33—38 Cuando SMi 2 es el donador de electrones, el primer intermedio reactivo es un cetilo de samario formado por reducción de un electrón del grupo carbonilo. 1 En la reacción mostrada en el Esquema 9, el átomo de carbono en el que está centrado el radical en el cetil de samario 31 se suma internamente al doble enlace carbono-nitrógeno que conduce al producto cíclico 32. Cuando está presente un exceso de SMi 2, la transferencia de electrones a 32 provoca la escisión del enlace N-O y el reemplazo final del grupo O-bencilo unido al nitrógeno con un átomo de hidrógeno (Esquema 9). 36 Ciclización similar se ha observado cuando el radical tri- n-butilestaño reacciona con un éter oxima que contiene un grupo aldehído o cetógeno. 39
D. Ésteres de oxima como fuentes radicales
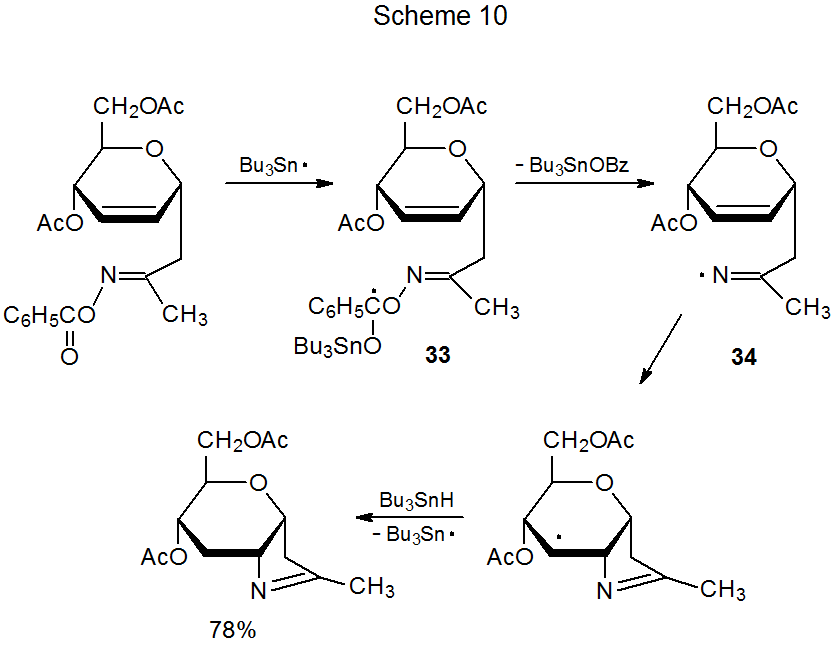
El evento característico de las reacciones de ciclación discutidas hasta ahora es un radical centrado en carbono que se agrega a un doble enlace carbono-nitrógeno en un éter de oxima. La mayoría de las reacciones formadoras de anillos que involucran derivados de oxima tienen lugar de esta manera, pero se produce un cambio en la reacción mostrada en el Esquema 10, donde el radical de adición (34) se genera a partir de una O-benzoiloxima. 40 En general, agregar Bu 3 Sn· a un grupo O-benzoílo es un comienzo poco propicio para una reacción radical porque dicha adición se invierte rápidamente, casi siempre antes de que pueda ocurrir cualquier otra reacción. Es posible una reacción adicional en este caso (Esquema 10) debido principalmente a la rápida fragmentación del débil enlace N-O en el radical 33 inicialmente formado. 40
E. Síntesis de oxima
Las oximas pueden ser los productos de la reacción radical. 41,42 Cuando se fotoliza el derivado D- glucopiranosilo que contiene cobalto 35, el radical producido se suma a una molécula de óxido nítrico para dar un compuesto nitroso. Este compuesto luego se tautomeriza para formar la oxima 36 correspondiente (eq 11). 41
.png)