II. Definición de características de las reacciones de adición de radicales
- Page ID
- 80008
A. Mecanismos de reacción
La adición de radicales puede tener lugar por reacción en cadena o no en cadena. Para cada uno de estos hay dos variaciones sobre el mecanismo básico de reacción. Para las reacciones en cadena (Esquemas 1 y 2) cada variación tiene un tipo diferente de etapa de transferencia de cadena. Para las reacciones no en cadena, ambos mecanismos implican transferencia de electrones. Se diferencian en que la transferencia es de (Esquema 3) o a (Esquema 4) un complejo organometálico.
1. Reacciones en Cadena
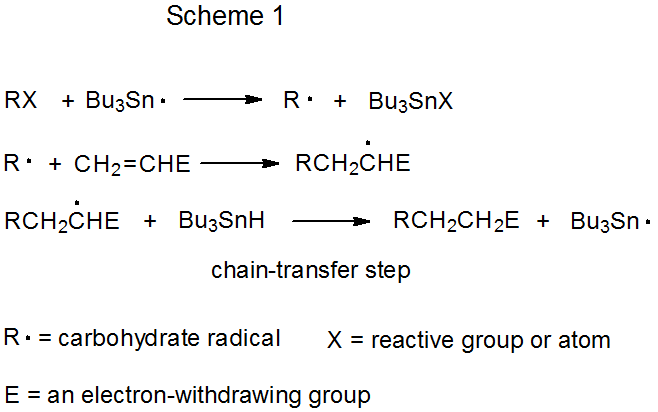
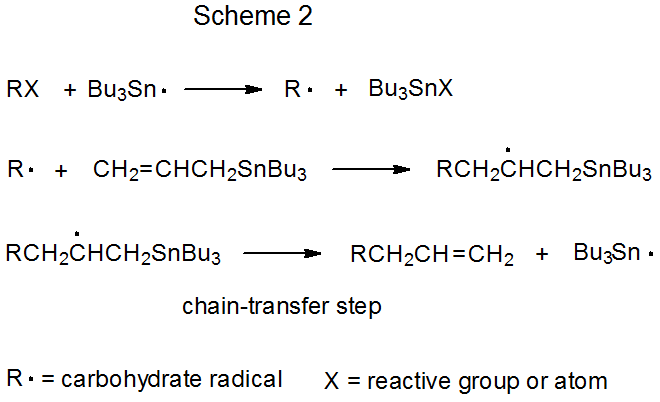
Ambos mecanismos para la adición de radicales por reacción en cadena tienen una fase de propagación que comienza con la abstracción de grupos o átomos (Esquemas 1 y 2). En cada uno de estos mecanismos Bu 3 Sn· se muestra como el radical de abstracción, aunque otros radicales [e.g., (Me 3 Si) 3 Si·] son capaces de llenar este papel. La diferencia definitoria entre estos dos mecanismos es el paso de transferencia de cadena. En la reacción mostrada en el Esquema 1 es bimolecular, y en que en el Esquema 2 es unimolecular.
a. Transferencia de Cadena Bimolecular
La característica distintiva de una reacción en cadena que tiene lugar por transferencia de cadena bimolecular es una reacción elemental entre un radical y un nonradical que termina una secuencia de propagación y crea el radical que inicia una nueva secuencia. En la reacción mostrada en el Esquema 1, la transferencia de cadena ocurre cuando el radical aducto extrae un átomo de hidrógeno de Bu 3 SnH para formar el producto de adición RCH 2 CH 2 E y generar el radical portador de cadena Bu 3 Sn·.
b. Transferencia de Cadena Unimolecular
La transferencia de cadena unimolecular describe una reacción en la que la etapa de transferencia de cadena en una secuencia de propagación es una reacción elemental con un solo reactivo. Los pasos de propagación para una reacción de transferencia de cadena típica, unimolecular, se muestran en el Esquema 2, donde la etapa de transferencia es una β-fragmentación que produce un compuesto insaturado y un radical portador de cadena.
2. Reacciones no en cadena
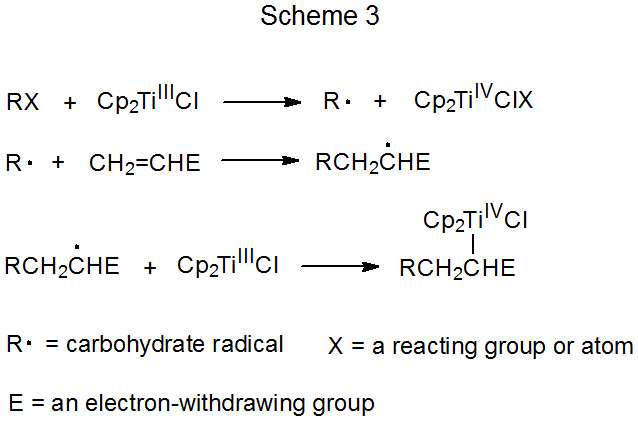
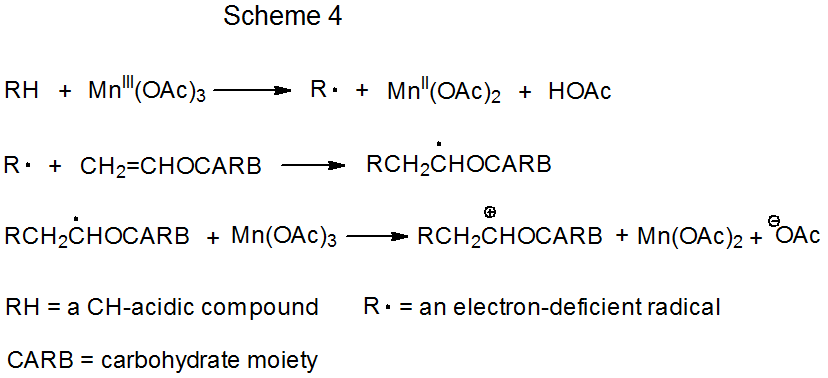
Los esquemas 3 y 4 describen cada uno un mecanismo para una reacción que no tiene ciclo de repetición. En el primero de estos (Esquema 3) el radical carbohidrato R· se forma por transferencia electrónica de un complejo de metal de transición (Cp 2 TiCl) a un derivado de carbohidrato. (Cp 2 TiCl es uno de varios complejos de metales de transición que se sabe que funcionan como donador de electrones en este tipo de reacción). La reacción radical termina cuando una segunda molécula de Cp 2 TiCl se combina con un radical aducto para producir un producto carbometálico inestable. Los productos de este tipo experimentan una rápida reacción no radical (por ejemplo, eliminación de los elementos de Cp 2 TiClH para formar un doble enlace).
En el segundo tipo de reacción no en cadena (Esquema 4) la formación de radicales ocurre cuando el complejo de metal de transición dona un electrón a un compuesto CH-ácido. La fase radical de la reacción termina con una segunda transferencia de electrones, una que produce un carbocatión. Este catión experimenta una reacción rápida, no radical, como la captura por una molécula de disolvente.
B. Selectividad en las reacciones de adición
1. Quimioselectividad
La quimioselectividad es de consecuencia en dos etapas en una reacción de adición de radicales. El primero es en la etapa de formación de radicales, donde la selectividad está determinada por la reactividad de los grupos funcionales presentes en una molécula de sustrato. La formación del radical deseado se logra en esta etapa asegurando que el precursor radical tenga el sustituyente más reactivo unido al átomo de carbono donde se va a ubicar el centro radical. El siguiente lugar en el que la selectividad potencialmente es significativa es durante la adición radical al enlace múltiple. La quimioselectividad es significativa en este momento si hay dos o más enlaces múltiples a los que puede ocurrir la adición.
2. Regioselectividad
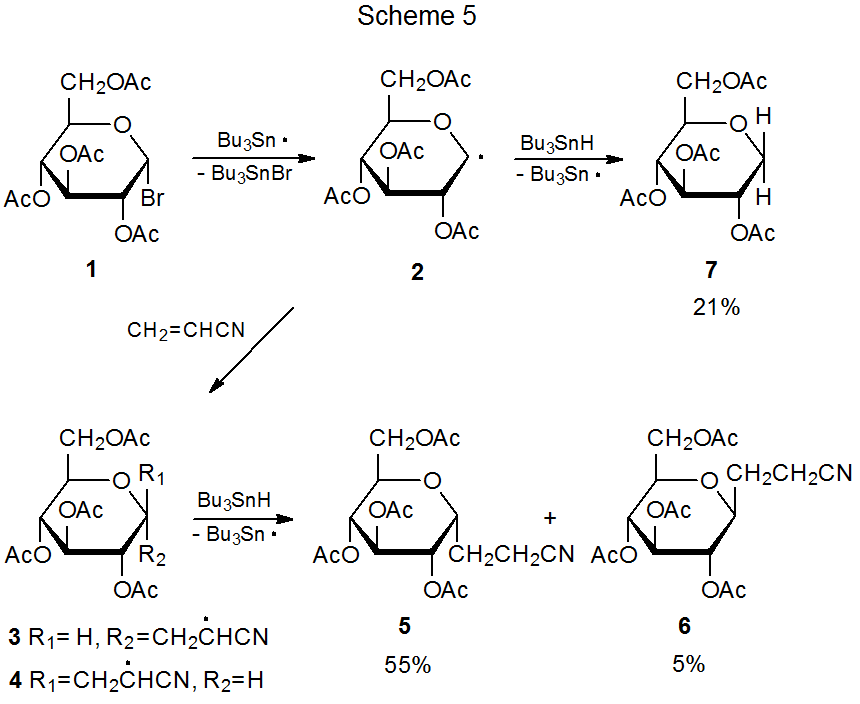
La mayoría de las reacciones de adición radical que involucran carbohidratos son regioespecíficas. La adición ocurre exclusivamente en el átomo de carbono menos sustituido en un enlace múltiple. La forma en que el radical piranos-1-ilo 2 se añade al acrilonitrilo es típica (Esquema 5). 1,2 Las reacciones de las Tablas 1 y 2 documentan una regioespecificidad similar en la adición de 2 a otros compuestos insaturados. Los datos de las Tablas 3-5 muestran que las reacciones de otros radicales piranos-1-ilo presentan una reactividad similar. (Todas estas tablas se encuentran al final de este capítulo.)
Tanto los efectos estéricos como los polares tienen un papel en la determinación de la regioespecificidad en las reacciones de adición. Los efectos estéricos se vuelven progresivamente más importantes a medida que aumenta el tamaño efectivo de los sustituyentes cerca de un enlace múltiple en un compuesto insaturado y a medida que aumenta el tamaño efectivo del radical agregado. Los efectos polares se ejercen cada vez que un radical nucleófilo se agrega a un enlace múltiple deficiente en electrones, o un radical electrófilo se agrega a un enlace múltiple rico en electrones. El alcance de la influencia de cada efecto depende del punto en el que se alcanza el estado de transición a medida que avanza una reacción. Por ejemplo, debido a que la adición de un radical nucleófilo a un enlace múltiple deficiente en electrones (el tipo más común de reacción de adición) es exotérmica, dicha reacción debería tener un estado de transición temprano (similar al reactivo) 11 con mínima formación de nuevos enlaces. Cuando hay poca formación de nuevos enlaces en el estado de transición, hay una menor oportunidad para que las interacciones estéricas afecten la regioselectividad. La situación con efectos polares es diferente porque, como se describe en el párrafo siguiente, pueden ejercer un considerable control regioselectivo en una reacción que tiene un estado de transición temprana.
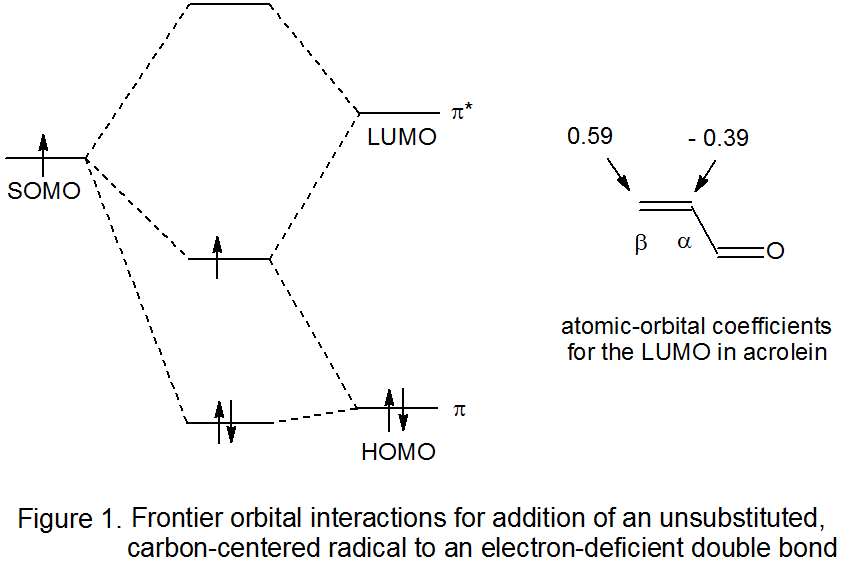
Para una reacción con un estado de transición temprana, los orbitales moleculares en los reactivos no cambian mucho para cuando se alcanza el estado de transición. En tal situación, las interacciones frontera-orbital son útiles no solo para comprender por qué se produce una reacción, sino también para explicar la regioselectividad de reacción. Brevemente, la reacción ocurre fácilmente porque el SOMO del radical carbohidrato y el LUMO del reactivo insaturado están lo suficientemente cerca energéticamente para una interacción estabilizadora significativa entre los dos en el estado de transición (Figura 1). 12,13 La extensión del enlace temprano entre un radical y los átomos de carbono en un enlace múltiple reactivo es una función de la magnitud del coeficiente orbital atómico en cada átomo de carbono en el LUMO del reactivo insaturado. La unión de un sustituyente aceptor de electrones o donador de electrones a un enlace múltiple hace que estos coeficientes sean bastante desiguales, situación que conduce a una reacción regioespecífica. Para un enlace múltiple polarizado como el que se encuentra en un compuesto de nitrilo o carbonilo α, β-insaturado, la adición regioespecífica al átomo de carbono β refleja la mayor magnitud del coeficiente orbital atómico en este átomo en el LUMO cuando se compara con la magnitud del coeficiente en el átomo de carbono α (Figura 1).
3. Estereoselectividad
Como se describe en las siguientes secciones, la estereoselectividad en las reacciones de adición de radicales está determinada por una combinación de efectos estéricos y estereoelectrónicos. El efecto estereoelectrónico de importancia primaria es el efecto anomérico cinético. Los efectos estéricos que tienen un impacto en la estereoselectividad de reacción tienen varios nombres, pero todos dependen de interacciones estéricas que favorecen un enfoque particular de un compuesto insaturado o una transferencia de átomo de hidrógeno a un centro radical.
a. El Efecto Anomérico Cinético
La reacción entre el radical piranos-1-ilo 2 y un compuesto insaturado con doble enlace deficiente en electrones es altamente estereoselectiva (Esquema 5). La reacción preferencial en la cara α del anillo piranoide en 2 se debe, en gran parte, al efecto anomérico cinético (discutido en el Capítulo 11 del Volumen I). La estereoselectividad en las reacciones de adición de 2 también se ve afectada por las condiciones de reacción. La información del Cuadro 1 incluye varios conjuntos de condiciones adecuadas para una reacción altamente estereoselectiva con poca reducción simple y competitiva.
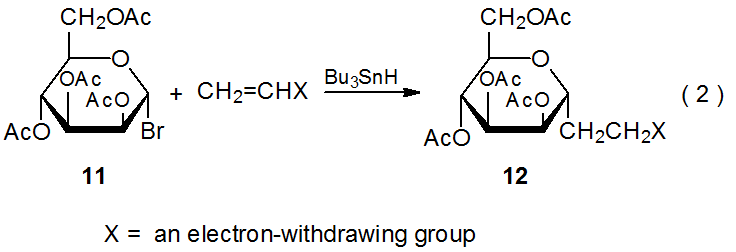
La selectividad observada en reacciones de 2 se extiende a otros radicales D-hexopiranos-1-ilo. Las reacciones de los bromidas D-galacto - y D-manopiranosilo 8 (eq 1) y 11 (eq 2), respectivamente, son al menos tan estereoselectivas como las del correspondiente bromuro de D-glucopiranosilo 1. Los datos de las Tablas 3 y 4 confirman que las reacciones de 8 y 11 ocurren preferentemente en la cara α del anillo piranoide.
.png)
.png)
b. Efectos Estéricos
(1). Blindaje de grupo
El blindaje grupal provoca una adición preferencial a la cara menos impedida de un sistema de anillos. En la reacción mostrada en la ecuación 3 el grupo 2,3- O-isopropilideno protege la cara α del radical centrada en C-4 en el anillo piranoide de la aproximación por el reactivo insaturado. 14
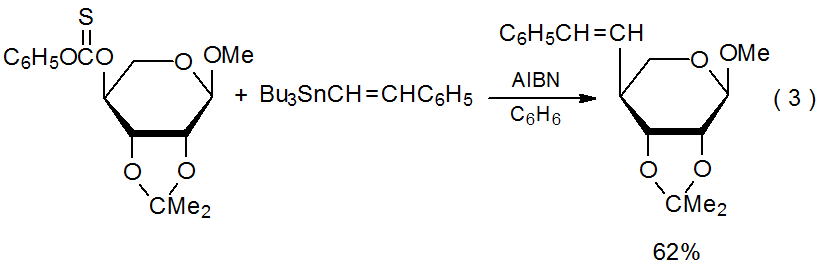.png)
(2). Tamaño del Reactivo Insaturado
La reacción mostrada en el Esquema 6 ilustra el efecto combinado del tamaño estérico del reactivo insaturado y el blindaje de grupo de un radical por sustituyentes en el anillo. 15 En esta reacción la cantidad de adición a la cara β mejor protegida del anillo piranoide en el radical 13 disminuye a medida que aumenta el tamaño efectivo del reactivo insaturado. Los datos del Esquema 6 muestran que, incluso en una reacción con un estado de transición temprana, los efectos estéricos pueden tener un papel significativo en la determinación de la estereoselectividad de reacción si estos efectos son lo suficientemente grandes.
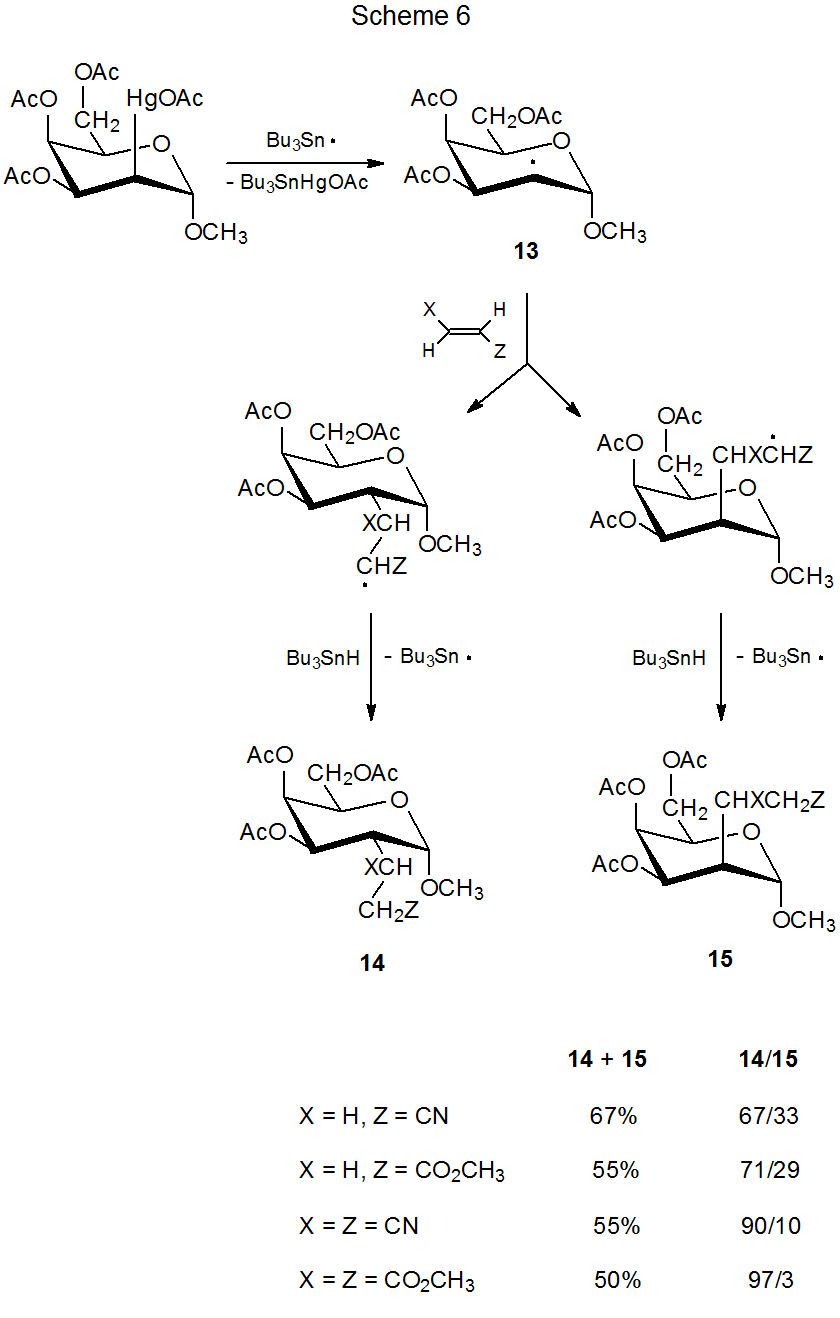
(3). Tamaño de la transferencia de átomos de hidrógeno
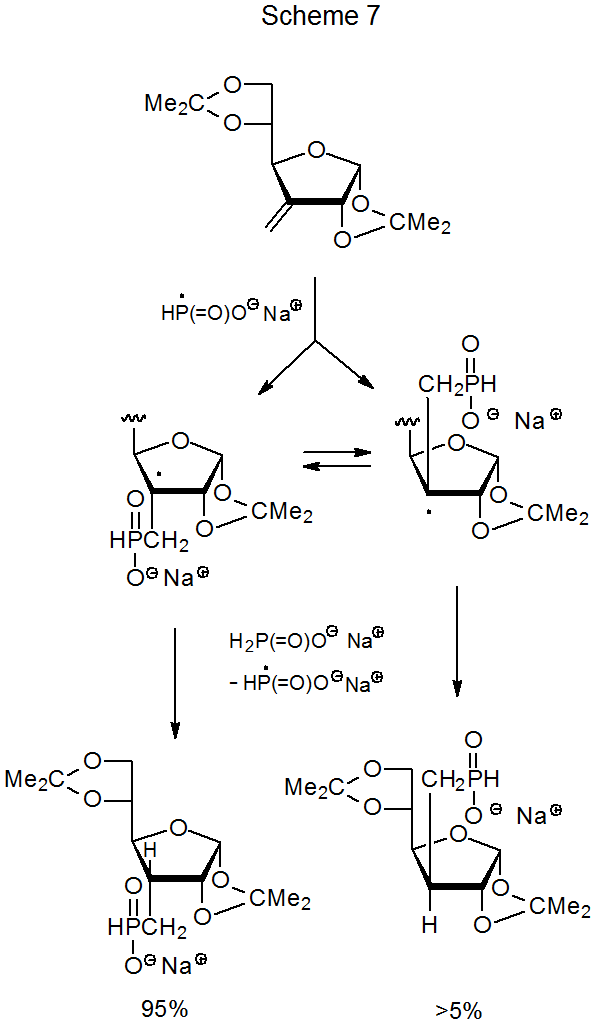
A veces, una reacción forma el menos estable de dos posibles estereoisómeros debido al enfoque restringido de una transferencia de átomos de hidrógeno a un centro radical en la etapa de formación del producto. En la reacción mostrada en el Esquema 7, por ejemplo, la aproximación menos impedida del hipofosfito de sodio a la cara β del anillo furanoide produce el estereoisómero menos estable en mayor el 95% de rendimiento. 16