1.3: Farmacocinética I
- Page ID
- 120371

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
- Describir los factores fisicoquímicos y fisiológicos que influyen en la absorción de fármacos de las vías de administración enteral y parenteral, su distribución dentro del cuerpo, y sus vías y mecanismos de eliminación.
- Explicar cómo la dosis, la biodisponibilidad, la tasa de absorción, el volumen aparente de distribución, el aclaramiento total y la vida media de eliminación afectan las concentraciones plasmáticas de un medicamento después de la administración de una sola dosis.
- Describir los factores que determinan el curso temporal de acumulación sistémica de un fármaco administrado por infusión o múltiples dosis.
Absorción de Drogas
- Transporte a través de membranas celulares
- Difusión pasiva
- Paso a través de la membrana celular lipídica por disolución en membrana; velocidad dependiente del gradiente de concentración y coeficiente de reparto lípido:agua del fármaco; velocidad marcadamente mayor para la forma sindicalizada de electrolito débil debido a su mayor lipofilia que la forma ionizada; obedece a la cinética de primer orden (tasa de el transporte es proporcional al gradiente de concentración en el sitio de transporte).
- Filtración a través de canales acuosos dentro de membranas y entre células.
- Transporte activo
- Paso facilitado por un mecanismo portador de membrana dependiente de energía tal que el transporte puede ocurrir contra un gradiente de concentración; los transportadores incluyen la familia de proteínas dependientes de ATP, tales como
- la p-glicoproteína multirresistente (sustratos anfipáticos catiónicos y neutros, 170 kD, producto génico mdr, sensible al verapamilo)
- las proteínas asociadas a la resistencia a múltiples fármacos (MRP1-6, sustratos aniónicos orgánicos, 190 kD, sensibles a probenecid).
- Presenta selectividad estructural, saturabilidad, competencia entre análogos estructurales y variantes genéticas.
- Sitios para fármacos en mucosa intestinal (célula a lumen), endotelio capilar de cerebro y testículos (célula a sangre), plexo coroideo (LCR a sangre), célula tubular renal proximal (sangre a orina), hepatocito (sangre a bilis), células tumorales (bomba de eflujo).
- Obedece la cinética de Michaelis-Menten: si la concentración de fármaco es lo suficientemente alta como para saturar el mecanismo portador, la cinética es de orden cero (la velocidad de transporte es constante).
- Paso facilitado por un mecanismo portador de membrana dependiente de energía tal que el transporte puede ocurrir contra un gradiente de concentración; los transportadores incluyen la familia de proteínas dependientes de ATP, tales como
- Endocitosis
- Paso a la célula dentro de la invaginación de la membrana.
- Mecanismo importante para partículas y compuestos de alto peso molecular, como las proteínas.
- Difusión pasiva
- Vías de Administración de Medicamentos
- Determinantes generales de la tasa de absorción
- Disolución en fluidos acuosos en el sitio de absorción, solubilidad lipídica, gradiente de concentración, flujo sanguíneo en el sitio de absorción, área superficial del sitio de absorción.
- Importancia del proceso de limitación de velocidad
- Oral (p.o.) Ingestión
- Vía conveniente para la administración de formulaciones sólidas así como líquidas.
- Las variables adicionales que pueden influir en la velocidad y el grado de absorción incluyen desintegración y disolución de sólidos, acidez del contenido gástrico, velocidad de vaciado gástrico, biotransformación intraluminal y mucosa por enzimas hospedadoras o bacterianas, contenido dietético y presencia de otros fármacos.
- Efecto de primer paso: el fármaco absorbido pasa a través de la circulación portal a través del hígado, lo que puede eliminar fracción sustancial y así disminuir la biodisponibilidad (porcentaje de dosis que alcanza la circulación sistémica).
- Inyección Parenteral
- Administración subcutánea (s.c.) e intramuscular (i.m.): absorción más extensa de moléculas polares de alto peso molecular que por vía oral, vía circulación linfática; la tasa de absorción puede ser manipulada por formulación, por ejemplo rápida a partir de solución acuosa, lenta desde suspensión o pellet sólido.
- Inyección intravenosa (i.v.): biodisponibilidad completa; fármacos administrados únicamente en solución estéril; importante cuando se requiere efecto inmediato; mayor riesgo de toxicidad.
- Inhalación pulmonar
- Rápida absorción de fármacos en forma gaseosa, vaporizada o aerosol.
- La absorción de partículas/aerosoles depende del tamaño de la partícula/gotita que influye en la profundidad de entrada en el árbol pulmonar; 1-5 uM partículas alcanzan alvéolo
- Aplicación Tópica
- Generalmente para efecto local; formulaciones de parche para efecto sistémico
- La absorción a través de la membrana mucosa puede ser rápida.
- La absorción a través de la piel generalmente es lenta; potenciada por el aumento de la lipofilia, por el daño al estrato córneo y por el aumento del flujo sanguíneo.
- Determinantes generales de la tasa de absorción
- Distribución de Medicamentos
- Diferencias tisulares en las tasas de absorción de fármacos.
- Flujo sanguíneo: la distribución ocurre más rápidamente en tejidos con flujo sanguíneo alto (pulmones, riñones, hígado, cerebro) y menos rápidamente en tejidos con bajo flujo (grasa).
- Permeabilidad capilar: la permeabilidad de los capilares es dependiente del tejido; tasas de distribución relativamente más lentas en el SNC debido a la estrecha unión entre las células endoteliales capilares, poros de membrana acuosa insignificantes, células gliales yuxtapuestas alrededor del endotelio y transportadores de eflujo en vascular endotelio (“barrera hematoencefálica”); capilares de hígado y riñón más porosos.
- Diferencias en las relaciones tejido/sangre en equilibrio
- Disolución de fármacos liposolubles en tejido adiposo
- Fijación de fármacos a sitios intracelulares
- Unión a proteínas plasmáticas; muchos fármacos se unen reversiblemente a la albúmina, α1-acidglicoproteína u otras proteínas en el plasma; el grado de unión depende de la afinidad, número de sitios de unión y concentraciones de fármaco; el fármaco unido a la albúmina no se filtra por glomérulo renal sino que puede ser aclarado por el túbulo renal proximal y el hígado ; la unión reduce el fármaco libre disponible para su distribución en el tejido; muchas interacciones farmacológicas basadas en el desplazamiento de los sitios de unión.
- Volumen aparente de distribución (V d)
- Compartimentos fluidos del sujeto de 70 kg en litros y como porcentaje del peso corporal: plasma 3 l (4%), agua extracelular 12 l (17%), agua corporal total 41 l (58%).
- Estimación de Vd a partir de la concentración plasmática extrapolada a “tiempo cero” (Co) después de la administración intravenosa:
\[\mathbf{V}_d={Dose \over \mathbf{C}_o}\]
- Predicción de Vd a partir de las características químicas del fármaco, por ejemplo, alta solubilidad lipídica, alta V d
- La vida media plasmática de un fármaco (el tiempo para reducir la concentración a la mitad) es directamente proporcional a Vd, e inversamente proporcional al aclaramiento total (Cl T); para un Cl T dado, cuanto mayor sea el V d, más largo será t 1/2:
\[\mathbf{t}_{1 \over 2}={ln2(\mathbf{V}_d )\over \mathbf{Cl}_T}\]
- Diferencias tisulares en las tasas de absorción de fármacos.
Eliminación de Drogas
- Liquidación Total (Cl T)
- Volumen de plasma completamente aclarado de fármaco por unidad de tiempo por todas las vías y mecanismos.
- Suma de los valores de aclaramiento para cada ruta, generalmente:
\[\mathbf{CL}_T={\mathbf {Cl}_{renal} + \mathbf{Cl}_{hepatic}}\]
- Si la capacidad intrínseca de un órgano para limpiar el fármaco es alta y excede el flujo plasmático a ese órgano, entonces el aclaramiento es igual al flujo plasmático y se ve alterado por cambios en el flujo plasmático.
- La vida media plasmática de un fármaco es inversamente proporcional al aclaramiento total, y directamente proporcional a Vd; para un Vd dado, cuanto mayor sea el aclaramiento total, más corta es la vida media.
- Biotransformación
- Eliminación del fármaco por modificación química de la molécula por reacción espontánea o (más habitualmente) catalizada enzimáticamente. El fármaco puede ser biotransformado por reacciones en varios sitios de la molécula.
- El (los) producto (s) puede (n) tener mayor, menor o cualitativamente diferente actividad farmacológica del compuesto parental. Un profármaco es inactivo y se biotransforma en un agente terapéutico. Los productos altamente reactivos como las quinonas o los epóxidos pueden causar necrosis tisular o daño al ADN.
- La velocidad de reacción depende de la estructura química y obedece a la cinética de Michaelis-Menten (generalmente de primer orden a concentraciones de fármaco terapéutico).
- La actividad enzimática generalmente mayor en el hígado; las enzimas en el órgano diana pueden ser responsables de la conversión del fármaco en metabolito terapéutico o tóxico; las enzimas en las bacterias intestinales pueden facilitar la circulación enterohepática de los conjugados farmacológicos excretados en la bilis.
- Fuentes de variación individual en las tasas de biotransformación: exposiciones químicas (fármacos, constituyentes y suplementos dietéticos, humo); genética; edad; enfermedad
- Principales vías de biotransformación hepática
- Fase I: a menudo primer paso en la biotransformación con formación de producto susceptible a reacción conjugativa de fase II
- Fase II: Acoplamiento del fármaco o su metabolito oxidado al agente conjugante endógeno derivado de fuentes de carbohidratos, proteínas o azufre; generalmente productos más solubles en agua y más fácilmente excretados en orina o bilis.
- Excreción
- Eliminación de fármaco por excreción inalterada en líquido corporal o aliento.
- Vías de excreción
- Orina: vía excretora cuantitativamente más importante para los fármacos no volátiles y sus metabolitos; la tasa de excreción depende de la tasa de filtración glomerular (fármaco no unido a proteínas plasmáticas), secreción activa tubular proximal y reabsorción pasiva
1) Determinación del aclaramiento renal (CLr), el volumen de plasma completamente aclarado de fármaco por unidad de tiempo (ml/min).
\[ {Cl}_R={{excreation\ rate\ i urine}\over plasma\ concentration }\]
Mida la cantidad de fármaco excretado en la orina durante un intervalo de tiempo t 1 a t2. Encontrar la concentración plasmática del fármaco en el punto medio del intervalo de tiempo, (t 1 + t 2) /2, interpolando en la gráfica ln C p vs t.
\[ {Cl}_R=[{ amount\ excreted \ from\ {t}_1\ to\ {t}_2\over ( {t}_2- {t}_1)}]\over {C}_p\ at {{( {t}_1 + {t}_2)}\over2}\]
2) El mecanismo de excreción renal se puede inferir comparando Cl R con el de un indicador de filtración glomerular (creatinina), es decir, mayor de 120 ml/min en sujetos de 70 kg indica secreción tubular y menor que eso indica reabsorción neta (si no hay unión plasmática); aclaramiento renal máximo = flujo plasmático renal (por ejemplo, ácido para-aminohipúrico, 650 ml/min en un sujeto de 70 kg).
3) Factores modificadores de Cl R: grado de unión a proteínas plasmáticas (el desplazamiento potencia la filtración glomerular), pH urinario (la reabsorción de fármacos con grupo ionizable depende del pH urinario; elevar el pH promueve la excreción de ácidos, perjudica la excreción de bases), enfermedad renal (aclaramiento de creatinina o su estimación a partir de creatinina sérica proporciona un indicador clínico útil de alteración de la función renal y es aproximadamente proporcional al aclaramiento renal farmacológico; el efecto de la insuficiencia renal sobre el aclaramiento total de un fármaco puede estimarse a partir del Cl CR y el aclaramiento no renal).
- Bilis: vía excretora cuantitativamente importante para los fármacos y sus metabolitos que son transportados activamente por los hepatocitos; una vez en el intestino delgado, los compuestos con suficiente lipofilia son reabsorbidos y limpiados nuevamente por el hígado (circulación enterohepática), sustancias más polares pueden ser biotransformadas por bacterias (por ejemplo, hidrólisis de conjugados de fármacos) y productos reabsorbidos; los fármacos y metabolitos no absorbidos se excretan en las heces.
- Vías menores: sudor, lágrimas, fluidos reproductivos, leche; difusión pasiva generalmente dependiente del pH de fármacos lipofílicos; puede ser de importancia toxicológica, por ejemplo, exposición de bebés a medicamentos en la leche.
- Orina: vía excretora cuantitativamente más importante para los fármacos no volátiles y sus metabolitos; la tasa de excreción depende de la tasa de filtración glomerular (fármaco no unido a proteínas plasmáticas), secreción activa tubular proximal y reabsorción pasiva
Curso temporal de las concentraciones plasmáticas
- Relación entre la concentración plasmática y el efecto del fármaco: concentración mínima efectiva, latencia, duración del efecto, tiempo y magnitud del efecto pico
- Evolución temporal de las concentraciones plasmáticas para una sola dosis
- Caso con Absorción Altamente Rápida Relativa a la Eliminación
- Modelo de compartimento único
1) Eliminación de primer orden: se supone que el fármaco se equilibra rápidamente en volumen de distribución; las concentraciones plasmáticas disminuyen según la cinética de primer orden; la tasa de eliminación del plasma es proporcional a la concentración plasmática, la fracción eliminada por unidad de tiempo es constante de velocidad de eliminación (k el).
\[{ {dC}_p\over dt}=- {k}_{el} {C}_p\]
\[ {C}_p= {C}_0e^{- {k}_{el}t}\]
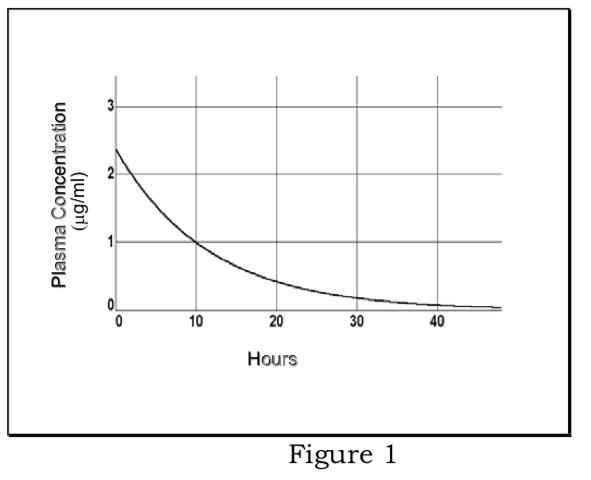
Determinación de la constante de velocidad de eliminación y semivida de eliminación:
\[ {lnC}_p=ln {C}_0- {k}_{el}t\]
La gráfica de ln C p vs. t es una línea recta con pendiente de -k el. La vida media plasmática (t 1/2 =.693/k el) es constante e independiente de la dosis.
Determinación del volumen aparente de distribución:
Extrapolación al tiempo cero de la línea de mejor ajuste para ln C p vs t datos; antilog de concentración de fármaco en el tiempo 0 designado como C 0. Entonces,
\[ {V}_d (in\ mls\ or\ liters)={Total\ Dose\over {C}_0}\]
Determinación del aclaramiento total:
Según las definiciones anteriores, el aclaramiento total es la masa de fármaco (Cp Vd) eliminada por unidad de tiempo dividida por la concentración plasmática; por lo tanto,
\[ {Cl}_T={( {k}_{el})( {C}_p\cdot {V}_d)\over {C}_p}={( {k}_{el})( {V}_d)}=[{0.693\over {t}_{1/2}}]( {V}_d)\]
Determinación del aclaramiento no renal (CLnR):
Si se determina el aclaramiento total y el aclaramiento renal a partir de muestras de plasma y orina como se describió anteriormente, entonces el aclaramiento por vías no renales (que incluye biotransformación) se puede estimar a partir de
\[ {Cl}_{NR}= {Cl}_T- {Cl}_R\]
2) Cinética de eliminación de orden cero: la tasa de eliminación es constante, t 1/2 es dependiente de la dosis (ejemplo: etanol).
3)
\[ {C}_p= {C}_0- {k}_0t\]
- Modelo multicompartimento
Distribución no instantánea de sangre a tejido dando como resultado una curva de concentración plasmática multiexponencial, la fase inicial refleja la distribución fuera del compartimento central en Vd total, la fase terminal refleja la eliminación.
\[ {C}_p=Ae^{-\alpha t}+Be^{-\beta t}\]
Donde α y β son constantes de velocidad híbridas que describen las 2 pendientes.
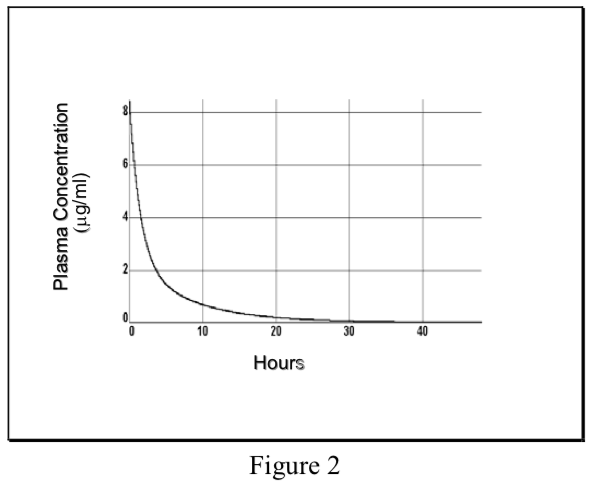
- Modelo de compartimento único
- Estuche con Absorción No Instantánea
- Cinética de absorción y eliminación de primer orden: determinación de semividas de absorción y eliminación
\[ {C}_p={ {k}_aFD\over {V}_d( {k}_a- {k}_{el})}[{e^{- {k}_{el}t}-e^{- {k}_at}}]\]
Tenga en cuenta que la pendiente terminal puede ser la constante de velocidad de eliminación, la constante de velocidad de absorción o un híbrido
Ver Katzung, Farmacología Básica y Clínica, 2001, p. 42
- La concentración plasmática máxima depende de las semividas de absorción y eliminación, el volumen de distribución, la dosis (D) y la fracción de dosis absorbida (F)
- \[AUC={F\cdot D\over {Cl}_T}\]
La fracción de dosis absorbida en la circulación sistémica (F) es la biodisponibilidad del producto farmacológico; determinada experimentalmente midiendo el AUC de la forma de dosificación del fármaco dada por una vía y comparándolo con el AUC de la misma dosis de fármaco en condiciones de absorción completa, es decir, administrado i.v.
- Cinética de absorción y eliminación de primer orden: determinación de semividas de absorción y eliminación
- Caso con Absorción Altamente Rápida Relativa a la Eliminación
- Efecto de las infusiones o dosis múltiples sobre el transcurso del tiempo de las concentraciones plasmáticas
- Cinética de Infusión
Un enfoque para mantener un nivel terapéutico deseado de un fármaco es administrar el agente por infusión intravenosa. La administración del fármaco puede controlarse mediante goteo regulado por gravedad del agente en tubos intravenosos o mediante el uso de una bomba de infusión.
- Cuando un fármaco se administra a una tasa de dosificación constante (DR) y su eliminación sigue una cinética de primer orden, la concentración de fármaco en el plasma aumenta exponencialmente y alcanza un nivel de estado estacionario o meseta (C ss).
\[ {C}_p(t)= {C}_{ss}(1-e^{ {-k}_{el}t})\]
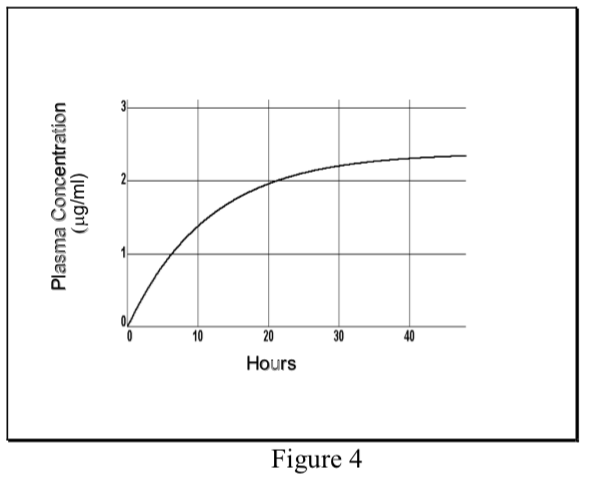
- En estado estacionario la TARIFA DE ENTRADA = TASA DE SALIDA. La tasa de entrada es DR, que puede expresarse como la dosis total (D) dividida por la duración de la infusión (T). La tasa de salida en el caso de eliminación de primer orden es la cantidad total de fármaco en el cuerpo (C ss Vd) multiplicada por la constante de velocidad de eliminación (k el).
\[DR= {C}_{ss}\cdot {V}_d\cdot{k}_{el}\]
Por lo tanto, la concentración plasmática en estado estacionario puede predecirse de la siguiente manera:
\[ {C}_{ss}=DR\over {V}_d {k}_{el}\]
Recuerde que el aclaramiento total equivale a la constante de velocidad de eliminación (k el) multiplicada por el volumen de distribución. Por lo tanto, la concentración plasmática en estado estacionario (C ss) es directamente proporcional a la tasa de entrada (DR) del fármaco e inversamente proporcional a su aclaramiento plasmático total (Cl T).
\[ {C}_{ss}={DR\over {Cl}_T}\]
- La tasa de logro del estado estacionario depende únicamente de la vida media de eliminación del fármaco. La mitad del nivel de C ss se logra en una t 1/2, y alrededor del 94% de C ss en cuatro t 1/2.
- Debido al retraso en lograr el estado estacionario cuando se administra una velocidad de infusión constante, se puede administrar una dosis de carga para lograr el efecto terapéutico deseado más rápidamente. La dosis de carga puede elegirse para producir la cantidad de fármaco en el cuerpo que eventualmente sería alcanzada por la infusión sola.
\[Loading\ dose= {C}_{ss}\cdot {V}_d\]
Al menos sobre una base teórica, la concentración plasmática alcanzará instantáneamente el nivel terapéutico y ese nivel se mantendrá. Obsérvese que el nivel de estado estacionario alcanzado con una infusión continua está determinado por la velocidad de infusión y no se ve afectado por el tamaño de la dosis de carga.
- Cuando un fármaco se administra a una tasa de dosificación constante (DR) y su eliminación sigue una cinética de primer orden, la concentración de fármaco en el plasma aumenta exponencialmente y alcanza un nivel de estado estacionario o meseta (C ss).
- Cinética de dosificación múltiple
- Comúnmente, los fármacos se administran repetidamente con el fin de mantener sus efectos terapéuticos. En el caso más simple, se administra una dosis de mantenimiento (D) en un intervalo de dosificación constante (\(\tau\)) — [tenga en cuenta que esto no es lo mismo que la constante de tiempo,\(\tau\)]. Dado que la vía de administración puede no ser i.v., la cantidad de fármaco que alcanza la circulación sistémica puede ser alguna fracción (F) de la dosis. Si la eliminación es por cinética de primer orden, finalmente se alcanza un estado estacionario. El Css “promedio” en estado estacionario es igual a la fracción absorbida por la tasa de dosificación dividida por el aclaramiento total, análogo al C ss de una infusión (ver arriba).
\ [{C} _ss\ promedio= {({F\ cdot D\ over\ tau})\ sobre {Cl} _T} $
- Sin embargo, en el caso de la dosificación repetitiva, a diferencia de una infusión, las concentraciones plasmáticas de fármaco fluctúan durante el intervalo de dosificación, dependiendo de la cinética de absorción y eliminación. El grado de fluctuación en la concentración plasmática durante un intervalo de dosificación aumenta al aumentar la dosis, el intervalo de dosificación, el aclaramiento y la tasa de absorción.
- Si un fármaco se administra i.v. (o donde la absorción es rápida y completa), la concentración plasmática máxima en estado estacionario (C maxss) en relación con el pico después
la primera dosis (C 0) depende de la relación entre la vida media de eliminación y el intervalo de dosificación (t 1/2/\(\tau\)).
- \[ {C}_{ {mass}_{ss}}={ {C}_0\over1-f}\]
f es la fracción de fármaco que queda al final de un intervalo de dosificación.
\[f=e^{ {-k}_{el}\cdot\tau}=e^{({-0.693\over {t}_{t/2}})\cdot\tau}=0.5^{\tau\over {t}_{1/2}}\]
Cada vez que se administra la dosis de mantenimiento D, la concentración plasmática aumenta de C min a C máx. La disminución de Cmax a Cmin se rige por la t 1/2, al igual que en una sola dosis. Estas relaciones se describen matemáticamente como:
\[ {C}_{ {min}_{ss}}+{D\over {V}_d}= {C}_{ {max}_{ss}}\]
\[{D\over {V}_d}={C}_0\]
\[ {C}_{ {min}_{ss}}+ {C}_0= {C}_{ {max}_{ss}}\]
\[ {lnC}_{ {min}_{ss}}= {lnC}_{ {max}_{ss}}-({0.693\over {t}_{t/2}}\tau)\]
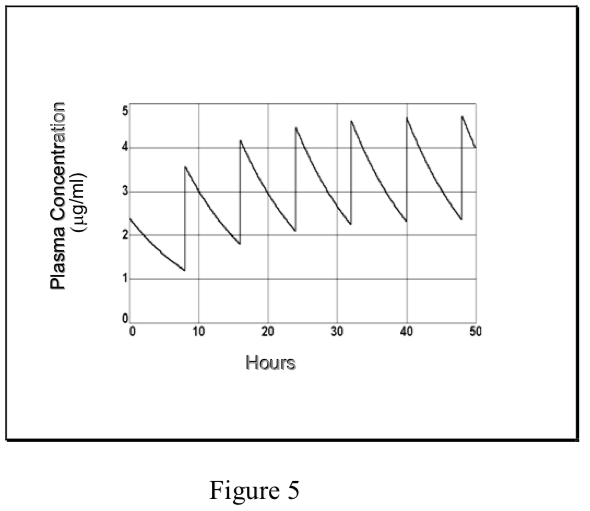
- La predicción de Cmax y Cmin en estado estacionario puede ser de gran importancia en los casos en los que se debe mantener la eficacia terapéutica minimizando el riesgo de efectos secundarios tóxicos. (Obsérvese que el “promedio” de Css descrito anteriormente se encuentra entre Cmaxss y Cminss, pero no es matemáticamente equivalente a su media aritmética o geométrica). La ventana terapéutica en un régimen de dosificación es el rango de concentraciones plasmáticas eficaces y no tóxicas que se encuentran entre Cmax ss y Cmin ss. Si se conocen estos, entonces el régimen de dosificación se determina de la siguiente manera:
\[Maintenance\ Dose = ( {C}_{ {max}_{ss}}- {C}_{ {min}_{ss}})\cdot {V}_d\]
\[Dosing \ interval \ (\tau)=[ln{ {C}_{ {max}_{ss}}\over {C}_{ {min}_{ss}}}][{ {t}_{t/2}\over0.693}]\]
- La velocidad de logro del estado estacionario está determinada por la vida media de eliminación (como con una infusión). Se puede usar una dosis de carga para alcanzar rápidamente concentraciones en estado estacionario; especialmente importante para medicamentos con vidas medias largas ya que el logro del estado estacionario es lento.
\[Loading\ dose = {C}_{ {max}_{ss}}\cdot {V}_d\]
- Comúnmente, los fármacos se administran repetidamente con el fin de mantener sus efectos terapéuticos. En el caso más simple, se administra una dosis de mantenimiento (D) en un intervalo de dosificación constante (\(\tau\)) — [tenga en cuenta que esto no es lo mismo que la constante de tiempo,\(\tau\)]. Dado que la vía de administración puede no ser i.v., la cantidad de fármaco que alcanza la circulación sistémica puede ser alguna fracción (F) de la dosis. Si la eliminación es por cinética de primer orden, finalmente se alcanza un estado estacionario. El Css “promedio” en estado estacionario es igual a la fracción absorbida por la tasa de dosificación dividida por el aclaramiento total, análogo al C ss de una infusión (ver arriba).
- Cinética de Infusión
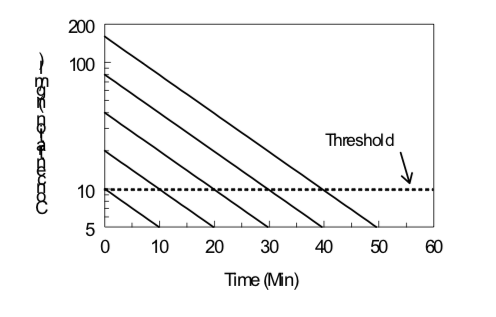
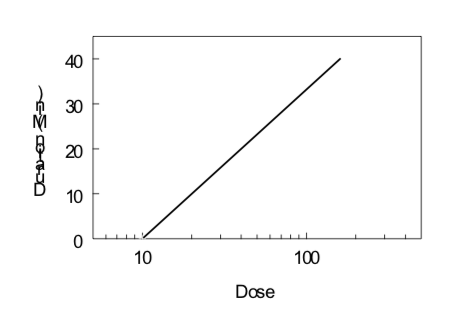
Tiempo-Curso del Efecto del Medicamento
Bajo ciertas condiciones (cinética de primer orden, efecto reversible, cinética de compartimento único, administración iv), la semivida de eliminación de un fármaco y su dosis umbral para un efecto particular pueden estimarse monitoreando el efecto del fármaco en función del tiempo después de la administración del fármaco. Los datos obtenidos a partir de varias dosis pueden ser evaluados examinando la duración de un determinado nivel de efecto en función del logaritmo de la dosis, como se ilustra a continuación. La pendiente es directamente proporcional a la vida media de eliminación; cuanto más pronunciada es la pendiente (es decir, aumentar la duración con un aumento de la dosis), mayor es la vida media de eliminación. La intercepción x indica el log de la dosis umbral; cuanto menor es la intercepción x, mayor es la potencia del fármaco.
\[Duration\ of\ Action ={ {t}_{1/2}\over0.301}(Log\ Dose\ - Log\ Threshold\ Dose)\]