
2.2: Alineación de secuencias
- Page ID
- 54667

La alineación de secuencias representa el método de comparación de dos o más cadenas genéticas, como ADN o ARN. Estas comparaciones ayudan con el descubrimiento de puntos en común genéticos y con el rastreo (implícito) de la evolución de las hebras. Hay dos tipos principales de alineación:
- Alineación global: un intento de alinear cada elemento de una cadena genética, más útil cuando las hebras genéticas consideradas son de aproximadamente el mismo tamaño. La alineación global también puede terminar en brechas.
- Alineación local: un intento de alinear regiones de secuencias que contienen motivos de secuencia similares dentro de un contexto más amplio.
Ejemplo de Alineación
Dentro de las secuencias génicas ortólogas, existen islas de conservación, o tramos relativamente grandes de nucleótidos que se conservan entre generaciones. Estas regiones conservadas suelen implicar elementos funcionales y viceversa. Como ejemplo, se consideró el alineamiento de la región intergénica Gal10-Gal1 para cuatro especies diferentes de levaduras, la primera alineación cruzada del genoma completo de especies (Figura 2.1). Al mirar este alineamiento, observamos que algunas áreas son más similares que otras, lo que sugiere que estas áreas se han conservado a través de la evolución. En particular, observamos algunos pequeños motivos conservados como CGG y CGC, que de hecho son elementos funcionales en la unión de Gal4 [8]. 2 Este ejemplo destaca cómo los datos evolutivos pueden ayudar a localizar áreas funcionales del genoma: los niveles de conservación por nucleótido denotan la importancia de cada nucleótido, y los exones se encuentran entre los elementos más conservados en el genoma.
Tenemos que ser cautelosos con nuestras interpretaciones, sin embargo, porque la conservación a veces ocurre por casualidad aleatoria. Para extraer información biológica precisa de alineaciones de secuencias tenemos que separar las firmas verdaderas del ruido. El enfoque más común de este problema implica modelar el proceso evolutivo. Mediante el uso de frecuencias de sustitución de codones conocidas y restricciones de estructura secundaria de ARN, por ejemplo, podemos calcular la probabilidad de que la evolución actuó para preservar una función biológica. Ver Capítulo?? para una discusión en profundidad sobre el modelado evolutivo y la conservación funcional en el contexto de la anotación genómica.
Resolución de alineación de secuencias
Los genomas cambian con el tiempo, y la escasez de genomas antiguos hace prácticamente imposible comparar los genomas de las especies vivas con los de sus antepasados. Así, nos limitamos a comparar solo los genomas de descendientes vivos. El objetivo del alineamiento de secuencias es inferir las 'operaciones de edición' que cambian un genoma observando solo estos puntos finales.
Debemos hacer algunas suposiciones a la hora de realizar el alineamiento de secuencias, aunque sólo sea porque debemos transformar un problema biológico en uno computacionalmente factible y requerimos un modelo con relativa simplicidad y trazabilidad. En la práctica, la evolución de la secuencia se debe principalmente a mutaciones, deleciones e inserciones de nucleótidos (Figura 2.2). Por lo tanto, nuestro modelo de alineación de secuencias solo considerará estas tres operaciones e ignorará otros eventos realistas que ocurren con menor probabilidad (por ejemplo, duplicaciones). 3
- Una mutación nucleotídica ocurre cuando algún nucleótido en una secuencia cambia a otro nucleótido durante el curso de la evolución.
- Una deleción de nucleótidos ocurre cuando se elimina algún nucleótido de una secuencia durante el curso de la evolución.
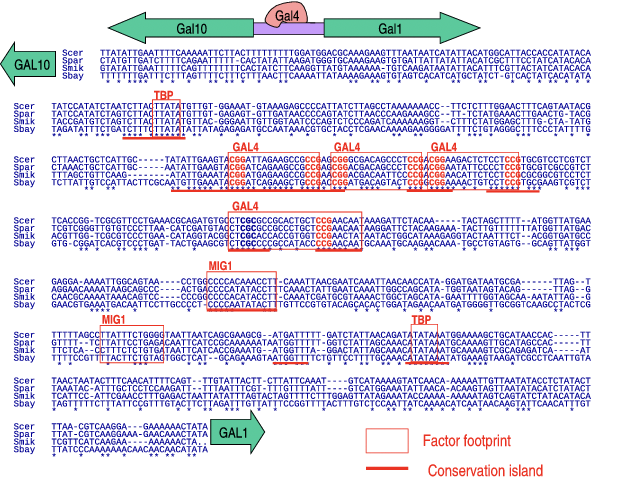
© fuente desconocida. Todos los derechos reservados. Este contenido está excluido de nuestra licencia Creative Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/. Figura 2.1: Alineación de secuencias de Gal10-Gal1 entre cuatro cepas de levadura. Los asteriscos marcan los nucleótidos conservados.
- Una inserción de nucleótidos ocurre cuando se agrega algún nucleótido a una secuencia durante el curso de la evolución.
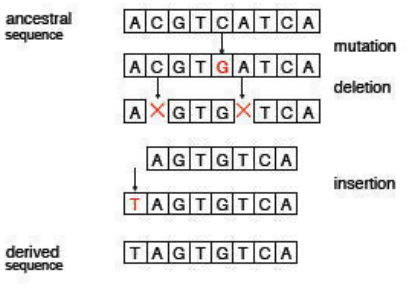
Tenga en cuenta que estos tres eventos son todos reversibles. Por ejemplo, si un nucleótido N muta en algún nucleótido M, también es posible que el nucleótido M pueda mutar al nucleótido N. De manera similar, si se elimina el nucleótido N, el evento puede revertirse si el nucleótido N es (re) insertado. Claramente, un evento de inserción se invierte por un evento de eliminación correspondiente.
Esta reversibilidad es parte de una suposición de diseño mayor: la reversibilidad en el tiempo. Específicamente, cualquier evento en nuestro modelo es reversible en el tiempo. Por ejemplo, una deleción de nucleótidos que avanza en el tiempo puede verse como una inserción de nucleótidos que retrocede en el tiempo. Esto es útil porque estaremos alineando secuencias que ambas existen en el presente. Para comparar la relación evolutiva, pensaremos en nosotros mismos siguiendo una secuencia hacia atrás en el tiempo a un ancestro común y luego continuaremos adelante en el tiempo a la otra secuencia. Al hacerlo, podemos evitar el problema de no tener una secuencia de nucleótidos ancestral.
Tenga en cuenta que la reversibilidad en el tiempo es útil para resolver algunos problemas biológicos, pero en realidad no se aplica a
sistemas biológicos. Por ejemplo, CpG4 4 puede emparejarse incorrectamente con un TpG o CpA durante la replicación del ADN, pero la operación inversa no puede ocurrir; de ahí que esta transformación no sea reversible en el tiempo. Para ser muy claros, la reversibilidad en el tiempo es simplemente una decisión de diseño en nuestro modelo; no es inherente a la biología 5.
También necesitamos alguna manera de evaluar nuestras alineaciones. Hay muchas secuencias posibles de eventos que podrían cambiar un genoma en otro. Quizás los más obvios minimizan el número de eventos (es decir, mutaciones, inserciones y deleciones) entre dos genomas, pero también son posibles secuencias de eventos en los que muchas inserciones son seguidas de deleciones correspondientes. Queremos establecer un criterio de optimalidad que nos permita escoger la “mejor” serie de eventos que describan los cambios entre genomas.
Elegimos invocar la navaja de Occam y seleccionar un método de máxima parsimonia como nuestro criterio de optimalidad. Es decir, en general, deseamos minimizar el número de eventos utilizados para explicar las diferencias entre dos secuencias de nucleótidos. En la práctica, encontramos que es más probable que ocurran mutaciones puntuales que inserciones y deleciones, y ciertas mutaciones son más probables que otras [11]. Nuestro método de parsimonia debe tener en cuenta estas y otras desigualdades a la hora de maximizar la parsimonia. Esto lleva a la idea de una matriz de sustitución y una penalización por gap, las cuales se desarrollan en los siguientes apartados. Tenga en cuenta que no fue necesario elegir un método de parsimonia máxima para nuestro criterio de optimalidad. Podríamos elegir un método probabilístico, por ejemplo usando Modelos Ocultos de Markov (HMM), que asignaría una medida de probabilidad sobre el espacio de posibles rutas de eventos y usaría otros métodos para evaluar alineaciones (por ejemplo, métodos bayesianos). Observe la dualidad entre estos dos enfoques: nuestro método de parsimonia máxima refleja la creencia de que los eventos de mutación tienen baja probabilidad, por lo que al buscar soluciones que minimicen el número de eventos estamos maximizando implícitamente su probabilidad.
2. Gal4 de hecho muestra una estructura particular, que comprende dos brazos que cada uno se une a la misma secuencia, en orden inverso.
3. Curiosamente, las decisiones de modelado tomadas para mejorar la trazabilidad no necesariamente resultan en una relevancia disminuida; por ejemplo, tener en cuenta la direccionalidad en el estudio de las inversiones cromosómicas produce soluciones polinomiales en tiempo para un problema de NP de otro modo. [6]
4. p denota la cadena principal de fosfato en una cadena de ADN
5. Este es un ejemplo donde entender la biología ayuda mucho al diseño, e ilustra el principio general de que el éxito en la biología computacional requiere un fuerte conocimiento de los fundamentos tanto de la CS como de la biología. Advertencia: los informáticos que ignoran la biología trabajarán demasiado duro.