
11.2: Regulación Postranscripcional
- Page ID
- 54965

Fundamentos de la traducción de proteínas
Para conocer los conceptos básicos de transcripción y traducción, consulte la Conferencia 1, secciones 4.3 - 4.5.

El código genético es casi universal.
|
FAQ P: ¿Por qué el código genético es tan similar entre los organismos? R: El material genómico no solo se transmite verticalmente (de los padres) sino también horizontalmente entre organismos. Esta interacción génica crea una presión evolutiva para un código genético universal. |
|
FAQ P: ¿Qué explica las ligeras diferencias en el código genético entre organismos? R: La llegada evolutiva tardía/temprana de aminoácidos puede explicar las diferencias. Además, ciertas especies (por ejemplo, bacterias en respiraderos de aguas profundas) tienen más recursos para sintetizar aminoácidos específicos, por lo que favorecerán a los del código genético. |
|
¿Sabías? La treonina y la alanina a menudo son intercambiadas accidentalmente por ARNt sintetasa porque se originaron a partir de un aminoácido. |
Traducción de medición
La eficiencia de la traducción se define como
\[T_{e f f}=\frac{[\mathrm{mRNA}]}{[\mathrm{protein}]}\nonumber\]
Nos interesa ver cuánto de nuestro ARNm se traduce en proteína, es decir, la eficiencia. Sin embargo, medir específicamente cuánto ARNm se convierte en proteína es una tarea difícil, una que requiere un poco de creatividad. Hay una variedad de formas de abordar este problema, pero cada una tiene sus propias caídas:
1. Medir los niveles de ARNm y proteínas directamente
Pitfall: No considera las tasas de síntesis y degradación. Este método mide los niveles de proteína para el ARNm 'viejo' ya que existe un retraso de tiempo de ARNm a proteína.
2. Usar drogas para inhibir la transcripción y la traducción. Dificultad: Las drogas tienen efectos secundarios que alteran la traducción
3. Fusión artificial de proteínas con etiquetas
Pitfall: Las etiquetas de proteínas pueden afectar la estabilidad de las proteínas
4. Marcador de pulso con nucleósidos o aminoácidos radiactivos (SILAC) **en uso hoy**
Dificultad: No ofrece información sobre los cambios dinámicos: es simplemente un snapshat de los niveles de ARNm y proteína resultantes después de X horas 193
Otra técnica común es usar el “perfil de ribosomas” para medir la traducción de proteínas en la resolución de subcodones. Esto se hace congelando ribosomas en el proceso de traducción y degradando las secuencias no protegidas por ribosomas. En este punto, las secuencias pueden ser reensambladas y la frecuencia con la que se traduce una región puede ser interpolada. La desventaja de usar estas huellas de ribosomas, para ver qué regiones se están traduciendo, es que se pierden regiones entre ribosomas. Esta técnica requiere una RNA-seq en paralelo.
La pregunta sigue siendo, ¿por qué es ventajoso el perfilado de ribosomas? Esta técnica es un mejor enfoque para medir la abundancia de proteínas ya que:
1. Es una mejor medida de la abundancia de proteínas
2. Es independiente de la degradación proteica (en comparación con la relación abundancia de proteínas/ARNm)
3. Nos permite medir tasas de traducción específicas de codones
Usando perfiles de ribosomas, es posible ver qué codón se está decodificando: esto se hace mapeando las huellas de ribosomas y luego descifrando el codón de traducción basado en la longitud de la huella. Podemos verificar nuestra predicción mapeando perfiles de codones traducidos basados en la periodicidad (tres bases en un codón). La técnica se puede mejorar aún más mediante el uso de fármacos anti-traducción como harringtonina y ciclohexamida. La ciclohexamida bloquea la elongación y la Harringtonina inhibe la iniciación. El último puede ser utilizado para encontrar los puntos de partida (qué genes están a punto de traducirse). La Figura 4 muestra los efectos de los fármacos sobre los perfiles de ribosomas.
Esta técnica tiene mucho más que ofrecer que simplemente cuantificar la traducción. El perfilado de ribosomas permite:
1. Predicción de isoformas alternativas (diferentes lugares donde puede comenzar la traducción) images/AltIsoforms.png

2. Predicción de ORF no identificados (marcos de lectura abiertos)
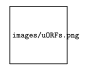
Figura 11.6: Perfil ribosómico cuando se usa harringtonina vs. sin fármaco. Los picos rojos previamente no identificados ORF.
3. Comparación de la traducción a través de diferentes condiciones ambientales

4. Comparando la traducción a través de las etapas
Así, vemos que el perfilado de ribosomas es una herramienta muy poderosa con mucho potencial para revelar información previamente esquiva sobre la traducción de un genoma.
Evolución de codones
Conceptos básicos
Algo que hay que dejar claro es que los codones no se utilizan con frecuencias iguales. De hecho, los codones que pueden considerarse óptimos difieren entre diferentes especies según la estabilidad del ARN, el sesgo de mutación específica de cadena, la eficacia transcripcional, la composición de GC, la hidropatía proteica y la eficiencia traduccional. Asimismo, los isoaceptores de ARNt no se utilizan con frecuencias iguales dentro y entre especies. La motivación para la siguiente sección es determinar cómo podemos medir este sesgo de codones.
Medidas del sesgo de codón
Existen algunos métodos para llevar a cabo esta tarea:
a) Calcular la frecuencia de codones óptimos, que se define como codones “óptimos”/suma de codones “óptimos” y “no óptimos”. Las limitaciones de este método son que esto requiere conocer qué codón es reconocido por cada ARNt y asume que la abundancia de ARNt está altamente correlacionada con el número de copias del gen del ARNt.
b) Calcular un índice de sesgo de codones. Esto mide la tasa de codones óptimos con respecto a los codones totales que codifican para ese mismo aminoácido. Sin embargo, en este caso el número de codones óptimos se normaliza con respecto al uso aleatorio esperado. CBI = (o opt − e rand)/(o tot − e rand). La limitación de este método es que requiere un conjunto de proteínas de referencia, como las proteínas ribosómicas altamente expresadas.
c) Calcular un índice de adaptación de codones. Esto mide la adaptabilidad relativa o desviación del codón us- age de un gen hacia el uso de codones de un conjunto de proteínas de referencia, es decir, genes altamente expresados. Se define como la media geométrica de los valores de adaptabilidad relativa, medidos como pesos asociados a cada codón sobre la longitud de la secuencia génica (medidos en codones). Cada peso se calcula como la relación entre la frecuencia observada de un codón dado y la frecuencia de su aminoácido correspondiente. La limitación a este enfoque es que requiere la definición de un conjunto de proteínas de referencia, tal como lo hizo el último método.
d) Calcular el número efectivo de codones. Esto mide el número total de codones diferentes utilizados en una secuencia, que mide el sesgo hacia el uso de un subconjunto más pequeño de codones, lejos del uso igual de codones sinónimos. Nc = 20 si solo se usa un codón por aminoácido, y Nc = 61 cuando todos los codones sinónimos posibles se usan por igual. Los pasos del proceso son calcular la homocigosidad para cada aminoácido estimada a partir de las frecuencias de codones al cuadrado, obtener el número efectivo de codones por aminoácido y calcular el número total de codones efectivos. Este método es ventajoso porque no requiere ningún conocimiento de emparejamiento ARNt-codón, y no requiere ningún conjunto de referencia. Sin embargo, está limitado en que no toma en cuenta el conjunto de ARNt.
e) Calcular el índice de adaptación del ARNt. Supongamos que el número de copias del gen de ARNt tiene una alta correlación positiva con abundancia de ARNt dentro de la célula. Esto luego mide qué tan bien se adapta un gen al acervo de ARNt.
Es importante distinguir entre cuándo usar cada índice. La situación en la que un determinado índice es favorable está muy basada en el contexto, por lo que a menudo es preferible utilizar un índice por encima de todos los demás cuando la situación lo requiere. Al elegir cuidadosamente un índice, se puede descubrir información sobre la frecuencia con la que un codón se traduce en un aminoácido.
Modificaciones de ARN
La historia se vuelve más complicada cuando consideramos modificaciones que pueden ocurrir al ARN. Por ejemplo, algunas modificaciones pueden expandir o restringir la capacidad de bamboleo del ARNt. Los ejemplos incluyen modificaciones de insosina y modificaciones de Xo5u. Estas modificaciones permiten a los ARNt decodificar un codón que antes no podían leer. Uno podría preguntarse por qué la modificación del ARN se seleccionó positivamente en el contexto de la evolución, y la razón es que esto permite aumentar la probabilidad de que exista un ARNt coincidente para decodificar un codón en un ambiente dado.
Ejemplos de aplicaciones
Hay algunas aplicaciones naturales que resultan de nuestra comprensión de la evolución de codones.
a) Optimización de codones para la expresión de proteínas heterólogas
b) Predicción de regiones codificantes y no codificantes de un genoma
c) Predicción de lectura de codones
d) Comprensión de cómo se decodifican los genes: estudio de patrones de sesgo de uso de codones
Regulación traslacional
Existen muchos medios conocidos de regulación a nivel postranscripcional. Estos incluyen la modulación de la disponibilidad de ARNt, cambios en el ARNm y elementos cis y trans-reguladores. Primero, la modulación de ARNt tiene un gran impacto. Cambios en los isoaceptores de ARNt, cambios en las modificaciones de ARNt y regulación en los niveles de aminoacilación de ARNt. Los cambios en el ARNm que afectan a la traducción incluyen cambios en la modificación del ARNm, la cola de poliA, el corte y empalme, el taponado y la localización del ARNm (importando y exportando desde el núcleo). Los elementos reguladores cis y trans incluyen interferencia de ARN (es decir, ARNip y miARN), eventos de cambio de marco y ribointerruptores. Además, ¡muchos elementos regulatorios aún están por descubrir!