
30.4: Rasgos mendelianos
- Page ID
- 54971
Mendel
Gregor Mendel identificó la primera evidencia de herencia en 1865 usando hibridación de plantas. Reconoció unidades discretas de herencia relacionadas con rasgos fenotípicos, y señaló que la variación en estas unidades, y por lo tanto variaciones en los fenotipos, fue transmisible a través de generaciones. Sin embargo, Mendel ignoró una discrepancia en sus datos: algunos pares de fenotipos no se transmitieron de forma independiente. Esto no se entendió hasta 1913, cuando el mapeo de enlaces mostró que los genes del mismo cromosoma se pasan en tándem a menos que ocurra un evento de cruce meiótico. Además, la distancia entre genes de interés describe la probabilidad de que ocurra un evento de recombinación entre los dos loci y, por lo tanto, la probabilidad de que los dos genes se hereden juntos (enlace).
Análisis de Vinculación
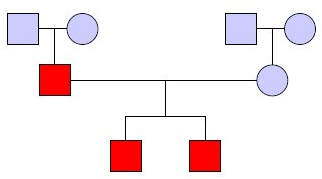
Históricamente, los investigadores han utilizado la idea de vinculación a través del análisis de ligamiento para determinar variantes genéticas que explican la variación fenotípica. El objetivo es determinar qué variantes contribuyen al patrón observado de variación fenotípica en un pedigrí. La Figura 30.3 muestra un pedigrí ejemplar en el que los cuadrados son individuos masculinos, los círculos son individuos femeninos, las parejas y las crías están conectadas, y los individuos en rojo tienen el rasgo de interés.
El análisis de ligamiento se basa en la percepción biológica de que las variantes genéticas no se heredan de forma independiente (como lo propone Mendel). En cambio, la recombinación meiótica ocurre un número limitado de veces (aproximadamente una vez por cromosoma), por lo que muchas variantes se cosegregan (se heredan juntas). Este fenómeno se conoce como desequilibrio de ligamiento (LD).
A medida que aumenta la distancia entre dos variantes, aumenta la probabilidad de que se produzca una recombinación entre ellas. Thomas Hunt Morgan y Alfred Sturtevant desarrollaron esta idea para producir mapas de ligamiento que no solo pudieran determinar el orden de los genes en un cromosoma, sino también sus distancias relativas entre sí. El Morgan es la unidad de distancia genética que propusieron; los loci separados por 1 centimórgano (cM) tienen 1 de cada 100 probabilidades de ser separados por una recombinación. Los loci no enlazados tienen 50% de probabilidad de ser separados por una recombinación (se separan si ocurre un número impar de recombinaciones entre ellos). Como generalmente no conocemos a priori qué variantes son causales, en su lugar utilizamos marcadores genéticos que capturan otras variantes debido a LD. En 1980, David Botstein propuso el uso de polimorfismos de un solo nucleótido (SNP), o mutaciones de una sola base, como marcadores genéticos en humanos [4]. Si un marcador particular está en LD con la variante causal real, entonces observaremos su patrón de herencia contribuyendo a la variación fenotípica en el pedigrí y puede estrechar nuestra búsqueda.
Los fundamentos estadísticos del análisis de vinculación se desarrollaron en la primera parte del siglo XX. Ronald Fisher propuso un modelo genético que pudiera conciliar la herencia mendeliana con fenotipos continuos como la altura [10]. Newton Morton desarrolló una prueba estadística llamada puntaje LOD (logaritmo de probabilidades) para probar la hipótesis de que los datos observados resultan de la vinculación [26]. La hipótesis nula de la prueba es que la fracción de recombinación (la probabilidad de que se produzca una recombinación entre dos marcadores adyacentes)\(\theta\) = 1/2 (sin vinculación) mientras que la hipótesis alternativa es que es una cantidad menor. La puntuación LOD es esencialmente una razón logarítmica de verosimilitud que captura esta prueba estadística:
\[\mathrm{LOD}=\frac{\log (\text { likelihood of disease given linkage })}{\log (\text { likelihood of disease given no linkage })}\nonumber\]
Los algoritmos para el análisis de ligamiento se desarrollaron en la última parte del siglo XX. Existen dos clases principales de análisis de ligamiento: paramétrico y no paramétrico [34]. El análisis de vinculación paramétrica se basa en un modelo (parámetros) de la herencia, frecuencias y penetrancia de una variante particular. Que F sea el conjunto de fundadores (ancestros originales) en el pedigrí, que gi sea el genotipo del individuo i,\(\Phi_{i}\) déjese
el fenotipo del individuo i, y dejar que f (i) y m (i) sean el padre y la madre del individuo i. Entonces, la probabilidad de observar los genotipos y fenotipos en el pedigrí es:
\[L=\sum_{g_{1}} \ldots \sum_{g_{n}} \prod_{i} \operatorname{Pr}\left(\Phi_{i} \mid g_{i}\right) \prod_{f \in F} \operatorname{Pr}\left(g_{f}\right) \prod_{i \notin F} \operatorname{Pr}\left(g_{i} \mid g_{f(i)}, g_{m(i)}\right)\nonumber\]
El tiempo requerido para calcular esta probabilidad es exponencial tanto en el número de marcadores considerados como en el número de individuos en el pedigrí. Sin embargo, Elston y Stewart dieron un algoritmo para calcularlo de manera más eficiente asumiendo que no hay endogamia en el pedigrí [8]. Su visión fue que condicionada a los genotipos parentales, las crías son condicionalmente independientes. En otras palabras, podemos tratar el pedigrí como una red bayesiana para calcular de manera más eficiente la distribución de probabilidad conjunta. Su algoritmo escala linealmente en el tamaño del pedigrí, pero exponencialmente en el número de marcadores.
Hay varios problemas con el análisis de vinculación paramétrica. Primero, los marcadores individuales pueden no ser informativos (dar información inequívoca sobre la herencia). Por ejemplo, los padres homocigotos o el error de genotipado podrían llevar a marcadores poco informativos. Para sortear esto, podríamos escribir más marcadores, pero el algoritmo no escala bien con el número de marcadores. En segundo lugar, es sencillo idear parámetros de modelo para un trastorno mendeliano. Sin embargo, hacer lo mismo con los trastornos no mendelianos no es trivial. Finalmente, las estimaciones de LD entre marcadores no se soportan inherentemente.
El análisis de ligamiento no paramétrico no requiere un modelo genético. En cambio, primero inferimos el patrón de herencia dados los genotipos y el pedigrí. Luego determinamos si el patrón de herencia puede explicar la variación fenotípica en el pedigrí.
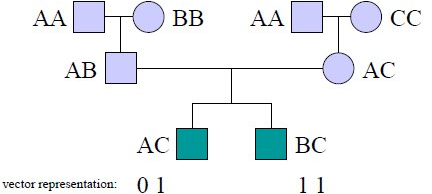
Lander y Green formularon un HMM para realizar la primera parte de este análisis [20]. Los estados de este HMM son vectores de herencia que especifican el resultado de cada meiosis en el pedigrí. Cada individuo está representado por 2 bits (uno para cada padre). El valor de cada bit es 0 o 1 dependiendo de cuál de los alelos grand-parental se hereda. La Figura 30.4 muestra un ejemplo de la representación de dos individuos en un vector de herencia.
Cada paso del HMM corresponde a un marcador; una transición en el HMM corresponde a algunos bits del vector de herencia cambiando. Esto significa que el alelo heredado de alguna meiosis cambió, es decir, que se produjo una recombinación. Las probabilidades de transición en el HMM son entonces una función de la fracción de recombinación entre marcadores adyacentes y la distancia de Hamming (el número de bits que difieren, o el número de recombinaciones) entre los dos estados. Podemos usar el algoritmo adelante/atrás para calcular las probabilidades posteriores en este HMM e inferir la probabilidad de cada patrón de herencia para cada marcador.
Este algoritmo escala linealmente en el número de marcadores, pero exponencialmente en el tamaño del pedigrí. El número de estados en el HMM es exponencial en la longitud del vector de herencia, que es lineal en el tamaño del pedigrí. En general, se sabe que el problema es NP-duro (a lo mejor de nuestro conocimiento, no podemos hacerlo mejor que un algoritmo que escala exponencialmente en la entrada) [28]. Sin embargo, el problema es importante no
nuestra licencia Creative Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
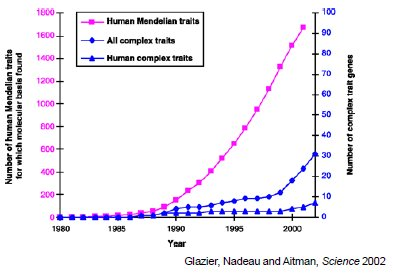
Fuente: Cristalero, Anne M., et al. “Encontrar genes que subyacen a rasgos complejos”. Ciencia 298, núm. 5602 (2002): 2345-9.
Figura 30.5: Descubrimiento de genes para diferentes tipos de enfermedad versus tiempo
sólo en este contexto, pero también en los contextos de inferencia o fase de haplotipos (asignación de alelos a cromosomas homólogos) e imputación de genotipos (inferir genotipos faltantes basados en genotipos conocidos). Ha habido muchas optimizaciones para que este análisis sea más manejable en la práctica [1, 11, 12, 15—18, 21, 23].
El análisis de ligamiento identifica una amplia región genómica que se correlaciona con el rasgo de interés. Para estrechar la región, podemos usar mapas genéticos de resolución fina de puntos de interrupción de recombinación. Luego podemos identificar el gen afectado y la mutación causal secuenciando la región y probando la función alterada.